


ホタル生物発光の量子化学計算:Gaussian を使った ONIOM TD-DFT 計算
- ホタルの生物発光 では、その発光色がどのように決まるのかについて、実験および理論の両面から、さまざまな研究が行われています
- 2010 年に Roland Lindh
らが発表した
論文
は、ホタル生物発光の量子化学計算に取り組むときのお手本としてオススメです
- この論文では、ゲンジボタルのルシフェラーゼ(発光酵素)とルシフェリン(発光基質)の複合体をモデル化して、QM/MM 法 と CASSCF/CASPT2 という手法を組み合わせることで、生物発光の発光色が決まるメカニズムを理論的に解析しています
- このノートでは、Lindh らの論文をお手本にしながら、代表的な量子化学計算アプリ Gaussian を使って、ONIOM 法 と TD-DFT 法 の組み合わせで、ホタル生物発光の量子化学計算を行う手順をまとめてみます
はじめに
略語
- 発光酵素:ゲンジボタルルシフェラーゼ (Luciola cruciata luciferase; LUC)
- 発光基質:オキシルシフェリン (Oxyluciferin; OLU)
- 補酵素:アデノシン一リン酸 (Adenosine monophosphate; AMP)
方針
- Nakatsu (2006) 論文
で報告されている複合体構造 (PDB ID: 2D1S
) を使います
- この構造では、発光基質アナログ (DLSA) が結合しています
- 今回は、LUC + OLU + AMP 複合体について、水和させた後、OLU を QM 領域、LUC と AMP を MM 領域に設定した ONIOM モデルをつくります
- DLSA を OLU + AMP に置き換えます
- 構造データに含まれる水分子は除去せず、その周りに水分子を追加します
手順
- まずは準備として、OLU と AMP の力場ファイルを準備します
- 次に、LUC に OLU と AMP が結合した複合体の構造データを作成します
- Amber を用いて、複合体に水分子を追加した水和構造を準備します
- Gaussian を使って、ホタル生物発光の量子化学計算を行います
準備
OLU と AMP の力場ファイル
OLU
- OLU の力場ファイルは、 こちら からダウンロードできます
立体構造の準備
- Protein Data Bank (PDB) から、OLU と LuLuc の複合体である 2D1R の構造データをダウンロードします
wget https://files.rcsb.org/download/2D1R.pdb- この構造データに含まれる OLU の立体構造を抽出します
grep "HETATM .* OLU " 2D1R.pdb > OLU_noH.pdb- 水素原子を追加した後、HETATM レコードの部分を抽出します
- ここでは、
Open Babel
(
obabel) を使っています
- ここでは、
Open Babel
(
obabel OLU_noH.pdb -O OLU_H.pdb -h; grep "HETATM" OLU_H.pdb > OLU.pdb- 出力された構造データを確認してみましょう
OLU.pdb
HETATM 1 O6' OLU A2001 16.670 26.100 10.887 1.00 0.00 O
HETATM 2 C4' OLU A2001 16.429 25.049 10.303 1.00 0.00 C
HETATM 3 N3' OLU A2001 17.314 24.123 9.956 1.00 0.00 N
HETATM 4 C2' OLU A2001 16.764 23.072 9.331 1.00 0.00 C
HETATM 5 S1' OLU A2001 15.145 23.066 9.100 1.00 0.00 S
HETATM 6 C5' OLU A2001 15.028 24.679 9.909 1.00 0.00 C
HETATM 7 C2 OLU A2001 17.572 21.905 8.870 1.00 0.00 C
HETATM 8 S1 OLU A2001 19.260 22.005 9.098 1.00 0.00 S
HETATM 9 C5 OLU A2001 19.363 20.481 8.469 1.00 0.00 C
HETATM 10 C6 OLU A2001 20.526 19.726 8.302 1.00 0.00 C
HETATM 11 C4 OLU A2001 18.079 19.867 8.068 1.00 0.00 C
HETATM 12 N3 OLU A2001 17.122 20.761 8.333 1.00 0.00 N
HETATM 13 C9 OLU A2001 18.054 18.591 7.519 1.00 0.00 C
HETATM 14 C8 OLU A2001 19.241 17.879 7.363 1.00 0.00 C
HETATM 15 C7 OLU A2001 20.463 18.435 7.744 1.00 0.00 C
HETATM 16 O7 OLU A2001 21.598 17.690 7.564 1.00 0.00 O
HETATM 17 H OLU A2001 14.403 24.616 10.775 1.00 0.00 H
HETATM 18 H OLU A2001 14.598 25.412 9.259 1.00 0.00 H
HETATM 19 H OLU A2001 21.437 20.117 8.589 1.00 0.00 H
HETATM 20 H OLU A2001 17.157 18.171 7.227 1.00 0.00 H
HETATM 21 H OLU A2001 19.216 16.929 6.961 1.00 0.00 H
HETATM 22 H OLU A2001 21.541 16.832 7.191 1.00 0.00 H- オキシ体は、ヒドロキシル (-OH) 基のプロトン(H+)が取れた状態です
- 今回の場合、該当するのは最後(22 番目)の原子ですね
- この原子を削除して、新しい構造データを作成します
- ここでは
sedコマンドを使いますが、エディタで修正してもオーケー
- ここでは
sed -i '22d' OLU.pdbOLU_H.pdb
HETATM 1 O6' OLU A2001 16.670 26.100 10.887 1.00 0.00 O
HETATM 2 C4' OLU A2001 16.429 25.049 10.303 1.00 0.00 C
HETATM 3 N3' OLU A2001 17.314 24.123 9.956 1.00 0.00 N
HETATM 4 C2' OLU A2001 16.764 23.072 9.331 1.00 0.00 C
HETATM 5 S1' OLU A2001 15.145 23.066 9.100 1.00 0.00 S
HETATM 6 C5' OLU A2001 15.028 24.679 9.909 1.00 0.00 C
HETATM 7 C2 OLU A2001 17.572 21.905 8.870 1.00 0.00 C
HETATM 8 S1 OLU A2001 19.260 22.005 9.098 1.00 0.00 S
HETATM 9 C5 OLU A2001 19.363 20.481 8.469 1.00 0.00 C
HETATM 10 C6 OLU A2001 20.526 19.726 8.302 1.00 0.00 C
HETATM 11 C4 OLU A2001 18.079 19.867 8.068 1.00 0.00 C
HETATM 12 N3 OLU A2001 17.122 20.761 8.333 1.00 0.00 N
HETATM 13 C9 OLU A2001 18.054 18.591 7.519 1.00 0.00 C
HETATM 14 C8 OLU A2001 19.241 17.879 7.363 1.00 0.00 C
HETATM 15 C7 OLU A2001 20.463 18.435 7.744 1.00 0.00 C
HETATM 16 O7 OLU A2001 21.598 17.690 7.564 1.00 0.00 O
HETATM 17 H OLU A2001 14.403 24.616 10.775 1.00 0.00 H
HETATM 18 H OLU A2001 14.598 25.412 9.259 1.00 0.00 H
HETATM 19 H OLU A2001 21.437 20.117 8.589 1.00 0.00 H
HETATM 20 H OLU A2001 17.157 18.171 7.227 1.00 0.00 H
HETATM 21 H OLU A2001 19.216 16.929 6.961 1.00 0.00 H- この PDB 形式の構造データを、xyz 座標形式に変換します
obabel OLU.pdb -O OLU.xyzOLU.xyz
21
OLU_new.pdb
O 16.67000 26.10000 10.88700
C 16.42900 25.04900 10.30300
N 17.31400 24.12300 9.95600
C 16.76400 23.07200 9.33100
S 15.14500 23.06600 9.10000
C 15.02800 24.67900 9.90900
C 17.57200 21.90500 8.87000
S 19.26000 22.00500 9.09800
C 19.36300 20.48100 8.46900
C 20.52600 19.72600 8.30200
C 18.07900 19.86700 8.06800
N 17.12200 20.76100 8.33300
C 18.05400 18.59100 7.51900
C 19.24100 17.87900 7.36300
C 20.46300 18.43500 7.74400
O 21.59800 17.69000 7.56400
H 14.40300 24.61600 10.77500
H 14.59800 25.41200 9.25900
H 21.43700 20.11700 8.58900
H 17.15700 18.17100 7.22700
H 19.21600 16.92900 6.96100量子化学計算
- 次に、準備した立体構造をもとに Gaussian を用いて OLU の構造最適化をおこない、この分子の部分電荷を推定します
- それでは、Gaussian の入力ファイルを作成しましょう
- OLU はプロトンが外れた状態なので、電荷が
-1であることに注意 - 部分電荷の推定には、 Merz-Kollman 法を使います
- OLU はプロトンが外れた状態なので、電荷が
gau.gjf
%Chk=gau.chk
#P B3LYP/6-31G(d) Opt=Tight
OLU
-1 1
O 16.67000 26.10000 10.88700
C 16.42900 25.04900 10.30300
N 17.31400 24.12300 9.95600
C 16.76400 23.07200 9.33100
S 15.14500 23.06600 9.10000
C 15.02800 24.67900 9.90900
C 17.57200 21.90500 8.87000
S 19.26000 22.00500 9.09800
C 19.36300 20.48100 8.46900
C 20.52600 19.72600 8.30200
C 18.07900 19.86700 8.06800
N 17.12200 20.76100 8.33300
C 18.05400 18.59100 7.51900
C 19.24100 17.87900 7.36300
C 20.46300 18.43500 7.74400
O 21.59800 17.69000 7.56400
H 14.40300 24.61600 10.77500
H 14.59800 25.41200 9.25900
H 21.43700 20.11700 8.58900
H 17.15700 18.17100 7.22700
H 19.21600 16.92900 6.96100
--Link1--
%Chk=gau.chk
#P B3LYP/6-31G(d) Geom=AllCheck Guess=(Read,Only) SCF=Tight
Pop=(Minimal,MK) IOp(6/33=2,6/41=10,6/42=17)- 上記の入力ファイルを使って、Gaussian を実行します
g16 < gau.gjf > gau.out- 分子研スパコンを使って計算する場合には、上記ではなく、
g16subコマンドを使います
g16sub gau.gjf- 計算が正常に終了したら、出力ファイル(
gau.out)とチェックポイントファイル(gau.chk)ができているはずです - 念のため、出力ファイルから最適化された構造を確認してみましょう
obabel gau.out -O gau.xyzgau.xyz
21
gau.out
O 4.89521 -1.52340 0.00000
C 3.97284 -0.72218 0.00000
N 2.63106 -1.03234 0.00000
C 1.84775 0.02257 -0.00001
S 2.64426 1.65053 -0.00000
C 4.25767 0.80878 0.00001
C 0.43879 -0.02536 -0.00001
S -0.45487 -1.58129 -0.00000
C -1.96541 -0.66923 -0.00000
C -3.25948 -1.12373 -0.00000
C -1.64011 0.74079 -0.00000
N -0.33733 1.05210 -0.00001
C -2.71224 1.68338 0.00000
C -4.00336 1.24454 0.00000
C -4.37000 -0.18246 0.00000
O -5.56600 -0.54524 0.00001
H 4.84369 1.06774 -0.88677
H 4.84367 1.06774 0.88680
H -3.50610 -2.18147 -0.00000
H -2.46402 2.74251 0.00000
H -4.83608 1.94387 0.00000力場の作成
- 量子化学計算で得られた結果をもとに、Amber の
antechamberツール を使って、OLU の力場をつくります - 一連の操作をまとめたスクリプト(
antechamber.sh)を準備しました
antechamber.sh
#!/bin/sh
lig=${1}
chg=${2}
echo Step 1:
# Generate an Antechamber file amb.ac from Gaussian output file gau.out
antechamber -fi gout -i gau.out -fo ac -o amb.ac -c resp -rn ${lig} -pf y
echo Step 2:
# Extract resp charge to a charge file amb.crg from amb.ac
antechamber -fi ac -i amb.ac -c wc -cf amb.crg -rn ${lig}
echo Step 3:
# Read in amb.crg and gau.pdb and generate an Anthechamber file amb.ac
antechamber -fi pdb -i ${lig}.pdb -fo ac -o amb.ac -c rc -cf amb.crg -nc ${chg} -rn ${lig} -pf y
echo Step 4:
# Judge the atom type using "atomtype". In this case, gaff atom type
# is generated amb.ac
atomtype -i amb.ac -o amb.ac -p gaff
echo Step 5:
# Generate residue topology file amb.prep using "prepgen". In this case
# a cartesian prep input file is generated with format tag (-f) of "car"
prepgen -i amb.ac -o amb.prep -f car -rn ${lig}
echo Step 6:
# Use parmchk to check the missing force field parameters and generate
# additional force field parameter file (frcmod) amb.frcmod
parmchk2 -i amb.prep -f prepc -o amb.frcmod
mv NEWPDB.PDB ${lig}.pdb
mv amb.prep ${lig}.prep
mv amb.frcmod ${lig}.frcmod
rm ATOMTYPE.INF PREP.INF QOUT esout punch qout amb.ac amb.crg- 分子研のスパコンを使う場合には、Amber の環境を
moduleコマンドで 読み込んでから、上記のスクリプトを実行します
module load amberantechamber.shスクリプトを実行します- 1つめの引数は、リガンドの名前であり、今回は
OLUにします - 2つめの引数は、電荷の設定であり、OLU の場合は
-1です
- 1つめの引数は、リガンドの名前であり、今回は
sh antechamber.sh OLU -1 | tee antechamber.log- 正常に終了したら、OLU 用に設定した力場がファイル(
OLU.frcmodなど)に出力されているはずです
OLU.frcmod
Remark line goes here
MASS
BOND
ANGLE
ne-c2-o 79.144 117.755 Calculated using o-c2-n2, penalty score= 2.8
ne-ce-ss 64.700 117.230 same as n2-ce-ss, penalty score= 2.8
cd-ce-ne 68.300 122.390 same as cc-ce-nf, penalty score= 1.5
cd-ce-ss 61.500 117.520 same as ca-ce-ss, penalty score= 2.0
ce-cd-ss 61.300 121.580 same as cf-cd-ss, penalty score= 0.8
DIHE
ss-cd-ce-ne 4 26.600 180.000 2.000 same as X -ce-cf-X , penalty score=136.0
nd-cd-ce-ne 4 26.600 180.000 2.000 same as X -ce-cf-X , penalty score=136.0
ne-ce-ss-c3 2 2.200 180.000 2.000 same as X -c2-ss-X , penalty score=237.0
cd-ce-ss-c3 1 1.100 180.000 -2.000 same as c2-c2-ss-c3
cd-ce-ss-c3 1 0.700 180.000 3.000 same as c2-c2-ss-c3, penalty score=324.0
ss-cd-ce-ss 4 26.600 180.000 2.000 same as X -ce-cf-X , penalty score=136.0
nd-cd-ce-ss 4 26.600 180.000 2.000 same as X -ce-cf-X , penalty score=136.0
ce-cd-ss-cc 2 2.200 180.000 2.000 same as X -c2-ss-X , penalty score=232.0
cd-cc-ss-cd 2 2.200 180.000 2.000 same as X -c2-ss-X , penalty score=232.0
cc-cc-ss-cd 2 2.200 180.000 2.000 same as X -c2-ss-X , penalty score=232.0
nd-cd-ss-cc 2 2.200 180.000 2.000 same as X -c2-ss-X , penalty score=232.0
IMPROPER
c3-ne-c2-o 1.1 180.0 2.0 Using the default value
cd-ne-ce-ss 1.1 180.0 2.0 Using the default value
ce-nd-cd-ss 1.1 180.0 2.0 Using the default value
cc-cd-cc-ss 1.1 180.0 2.0 Using the default value
c -cc-cd-ha 1.1 180.0 2.0 Same as X -X -ca-ha, penalty score= 38.9 (use general term))
cc-cc-cc-nd 1.1 180.0 2.0 Using the default value
cc-cd-cc-ha 1.1 180.0 2.0 Same as X -X -ca-ha, penalty score= 38.9 (use general term))
cd-cd-c -o 10.5 180.0 2.0 Using general improper torsional angle X- X- c- o, penalty score= 6.0)
NONBONOLU.prep
0 0 2
This is a remark line
molecule.res
OLU XYZ 0
CHANGE OMIT DU BEG
0.0000
1 DUMM DU M 999.000 999.0 -999.0 .00000
2 DUMM DU M 999.000 -999.0 999.0 .00000
3 DUMM DU M -999.000 999.0 999.0 .00000
4 O1 o M 4.895000 -1.523000 0.000000 -0.563608
5 C1 c2 M 3.973000 -0.722000 0.000000 0.616170
6 N1 ne E 2.631000 -1.032000 0.000000 0.163486
7 C3 c3 M 4.258000 0.809000 0.000000 -0.499580
8 H1 h1 E 4.844000 1.068000 -0.887000 0.231959
9 H2 h1 E 4.844000 1.068000 0.887000 -0.270522
10 S1 ss M 2.644000 1.651000 -0.000000 0.028279
11 C2 ce M 1.848000 0.023000 -0.000000 -0.168755
12 C4 cd M 0.439000 -0.025000 -0.000000 0.113282
13 S2 ss S -0.455000 -1.581000 -0.000000 -0.594986
14 C5 cc S -1.965000 -0.669000 -0.000000 0.037448
15 C6 cd S -3.259000 -1.124000 -0.000000 0.037448
16 H3 ha E -3.506000 -2.181000 -0.000000 0.200283
17 N2 nd M -0.337000 1.052000 -0.000000 -0.482352
18 C7 cc M -1.640000 0.741000 -0.000000 0.302676
19 C8 cc M -2.712000 1.683000 0.000000 -0.429314
20 H4 ha E -2.464000 2.743000 0.000000 -0.256735
21 C9 cd M -4.003000 1.245000 0.000000 -0.259188
22 H5 ha E -4.836000 1.944000 0.000000 0.533319
23 C10 c M -4.370000 -0.182000 0.000000 0.143215
24 O2 o M -5.566000 -0.545000 0.000000 0.117475
LOOP
C2 N1
C7 C5
C10 C6
IMPROPER
C3 N1 C1 O1
C4 N1 C2 S1
C2 N2 C4 S2
C7 C6 C5 S2
C10 C5 C6 H3
C5 C8 C7 N2
C7 C9 C8 H4
C10 C8 C9 H5
C6 C9 C10 O2
DONE
STOPOLU.pdb
ATOM 1 O1 OLU 1 4.895 -1.523 0.000 -0.563608
ATOM 2 C1 OLU 1 3.973 -0.722 0.000 0.616170
ATOM 3 N1 OLU 1 2.631 -1.032 0.000 0.163486
ATOM 4 C3 OLU 1 4.258 0.809 0.000 -0.499580
ATOM 5 H1 OLU 1 4.844 1.068 -0.887 0.231959
ATOM 6 H2 OLU 1 4.844 1.068 0.887 -0.270522
ATOM 7 S1 OLU 1 2.644 1.651 -0.000 0.028279
ATOM 8 C2 OLU 1 1.848 0.023 -0.000 -0.168755
ATOM 9 C4 OLU 1 0.439 -0.025 -0.000 0.113282
ATOM 10 S2 OLU 1 -0.455 -1.581 -0.000 -0.594986
ATOM 11 C5 OLU 1 -1.965 -0.669 -0.000 0.037448
ATOM 12 C6 OLU 1 -3.259 -1.124 -0.000 0.037448
ATOM 13 H3 OLU 1 -3.506 -2.181 -0.000 0.200283
ATOM 14 N2 OLU 1 -0.337 1.052 -0.000 -0.482352
ATOM 15 C7 OLU 1 -1.640 0.741 -0.000 0.302676
ATOM 16 C8 OLU 1 -2.712 1.683 0.000 -0.429314
ATOM 17 H4 OLU 1 -2.464 2.743 0.000 -0.256735
ATOM 18 C9 OLU 1 -4.003 1.245 0.000 -0.259188
ATOM 19 H5 OLU 1 -4.836 1.944 0.000 0.533319
ATOM 20 C10 OLU 1 -4.370 -0.182 0.000 0.143215
ATOM 21 O2 OLU 1 -5.566 -0.545 0.000 0.117475AMP
- AMP の力場ファイルは、 こちら からダウンロードできます
ADP の力場ファイルを入手
- Richard Bryce
らが公開している AMBER parameter database
から ADP の
prepファイル、 リン酸部分の力場パラメータが載っているfrcmodファイルをダウンロードします- 上記のデータベースを利用するので、論文を書くときには、 この文献 を引用するのを忘れないように
wget http://amber.manchester.ac.uk/cof/ADP.prepwget http://amber.manchester.ac.uk/cof/frcmod.phosADP.prep
0 0 2
R-ADENOSINE - with diphosphate linker
adp.db94
adp INT 1
CORRECT OMIT DU BEG
0.0
1 DUMM DU M 0 0 0 0.000 0.000 0.000 0.000
2 DUMM DU M 1 0 0 1.000 0.000 0.000 0.000
3 DUMM DU M 2 1 0 1.000 90.000 0.000 0.000
4 O1B O3 M 3 2 1 1.000 90.000 180.000 -0.9552
5 PB P M 4 3 2 1.434 90.000 180.000 1.3672
6 O2B O3 E 5 4 3 1.574 107.490 -79.441 -0.9552
7 O3B O3 E 5 4 3 1.518 118.007 165.990 -0.9552
8 O3A OS M 5 4 3 1.599 113.374 180.000 -0.6346
9 PA P M 6 5 4 1.646 130.619 36.624 1.4929
10 O1A O2 E 7 6 5 1.504 109.212 -90.234 -0.9474
11 O2A O2 E 7 6 5 1.526 108.570 40.323 -0.9474
12 O5* OS M 7 6 5 1.585 97.173 157.726 -0.6579
13 C5* CT M 8 7 6 1.445 122.292 -66.317 0.0558
14 H50 H1 E 9 8 7 1.059 109.484 27.531 0.0679
15 H51 H1 E 9 8 7 1.059 109.437 -92.456 0.0679
16 C4* CT M 9 8 7 1.477 113.286 147.538 0.1065
17 H40 H1 E 16 9 8 1.059 105.768 -179.178 0.1174
18 O4* OS S 16 13 9 1.482 106.235 -62.889 -0.3548
19 C1* CT B 18 16 13 1.391 107.688 128.804 0.0394
20 H10 H2 E 19 18 16 1.059 109.468 91.228 0.2007
21 N9 N* S 19 18 16 1.552 105.091 -143.300 -0.0251
22 C8 CK B 21 19 18 1.388 123.797 33.434 0.2006
23 H80 H5 E 22 21 19 1.078 131.047 -3.389 0.1553
24 N7 NB S 22 21 19 1.384 108.967 176.598 -0.6073
25 C5 CB S 24 22 21 1.395 103.021 0.702 0.0515
26 C6 CA B 25 24 22 1.412 128.288 178.252 0.7009
27 N6 N2 B 26 25 24 1.333 125.907 -3.791 -0.9019
28 H60 H E 27 26 25 1.010 120.010 2.829 0.4115
29 H61 H E 27 26 25 1.009 120.014 -177.167 0.4115
30 N1 NC S 26 25 24 1.343 113.319 178.851 -0.7615
31 C2 CQ B 30 26 25 1.340 123.433 -1.664 0.5875
32 H2 H5 E 31 30 26 1.077 120.022 -177.875 0.0473
33 N3 NC S 31 30 26 1.377 125.379 2.123 -0.6997
34 C4 CB E 33 31 30 1.300 108.242 -0.035 0.3053
35 C3* CT M 16 13 12 1.484 117.051 57.581 0.2022
36 H30 H1 E 35 16 13 1.059 117.939 24.820 0.0615
37 O3* OH S 35 16 13 1.407 108.509 149.908 -0.6541
38 H3* HO E 37 35 16 0.967 109.470 163.254 0.4376
39 C2* CT M 35 16 13 1.607 102.425 -96.715 0.0670
40 H20 H1 E 39 35 16 1.059 115.086 78.119 0.0972
41 O2* OH S 39 35 16 1.398 114.943 -153.294 -0.6139
42 H2* HO E 41 39 35 0.967 109.456 -41.679 0.4186
IMPROPER
C8 C4 N9 C1*
C6 H60 N6 H61
N7 N9 C8 H80
N1 N3 C2 H2
C5 N1 C6 N6
LOOP CLOSING EXPLICIT
C1* C2*
C4 C5
C4 N9
DONE
STOPfrcmod.phos
# Modifications to the AMBER94 force field for polyphosphates
MASS
CT 12.01
OS 16.00
P 30.97
H1 1.008
O2 16.00
O3 16.00
BOND
CT-H1 340.000 1.090 from amber98
CT-OS 320.000 1.410
OS-P 230.000 1.61000
O2-P 525.000 1.480
O3-P 525.000 1.480 by analogy to O2
ANGLE
H1-CT-OS 50.000 109.500
H1-CT-H1 35.000 109.500
CT-OS-P 100.000 120.500
O2-P -O2 140.000 119.900
O3-P -O3 140.000 119.90 by analogy to O2
OS-P -O2 100.000 108.230
OS-P -O3 100.000 108.23 by analogy to O2
OS-P -OS 45.000 102.600
P -OS-P 12.685 150.000 J Comp Chem 2003, 24, 1016-1025
DIHE
H1-CT-OS-P 3 0.105 000.000 3.000 J Comp Chem 2003, 24, 1016-1025
O2-P -OS-CT 2 1.179 000.000 -3.000 J Comp Chem 2003, 24, 1016-1025
O2-P -OS-CT 2 -0.812 000.000 2.000 J Comp Chem 2003, 24, 1016-1025
CT-OS-P -OS 1 -1.560 0.0 1.0 J Comp Chem 2003, 24, 1016-1025
O2-P -OS-P 2 -0.709 0.0 2.0 J Comp Chem 2003, 24, 1016-1025
O3-P -OS-P 3 -0.255 0.0 3.0 J Comp Chem 2003, 24, 1016-1025
P -OS-P -OS 1 0.897 0.00 1.0 J Comp Chem 2003, 24, 1016-1025
NONBON
H1 1.3870 0.0157 Veenstra et al JCC,8,(1992),963
O2 1.6612 0.2100 OPLS
O3 1.6612 0.2100 OPLS - by analogy to O2
CT 1.9080 0.1094 Spellmeyer
P 2.1000 0.2000 JCC,7,(1986),230;
OS 1.6837 0.1700 OPLS etherADP の prep ファイルを書き換え
ADP.prepを AMP 用に書き換えます(注意:かなり面倒です 😭)ADP.prepファイルから、PA、O1A、O2A、O5*を削除O1B→O1P,PB→P,O2B→O2P,O3B→O3P,O3A→O5'に変更- 原子名に付いているアスタリスク(
*)をプライム(')に置き換え - 原子の番号が順番通りになるように修正します
- 上記に対応し、リンクの部分にある原子番号(例:
M 3 2 1の数字)も修正する必要があります(これが面倒 😭)- GaussView などで分子構造を確認しながら修正することをお勧めします
- Lindh らの 論文 の SI に記載されている値を使って、リン酸部位の部分電荷を修正
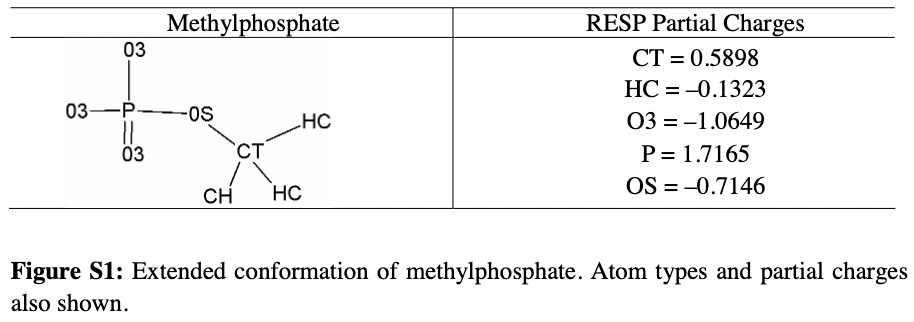
- 上記の修正の結果、AMP 用に書き換えたファイル(
AMP.prep)は下記のようになります
AMP.prep
0 0 2
R-ADENOSINE - with monophosphate linker
amp.db94
AMP INT 1
CORRECT OMIT DU BEG
0.0
1 DUMM DU M 0 0 0 0.000 0.000 0.000 0.000
2 DUMM DU M 1 0 0 1.000 0.000 0.000 0.000
3 DUMM DU M 2 1 0 1.000 90.000 0.000 0.000
4 O1P O3 M 3 2 1 1.000 90.000 180.000 -1.0649
5 P P M 4 3 2 1.434 90.000 180.000 1.7165
6 O2P O3 E 5 4 3 1.574 107.490 -79.441 -1.0649
7 O3P O3 E 5 4 3 1.518 118.007 165.990 -1.0649
8 O5' OS M 5 4 3 1.599 113.374 180.000 -0.7146
9 C5' CT M 8 7 6 1.445 122.292 -66.317 0.5898
10 H50 H1 E 9 8 7 1.059 109.484 27.531 -0.1323
11 H51 H1 E 9 8 7 1.059 109.437 -92.456 -0.1323
12 C4' CT M 9 8 7 1.477 113.286 147.538 0.1065
13 H40 H1 E 12 9 8 1.059 105.768 -179.178 0.1174
14 O4' OS S 12 11 9 1.482 106.235 -62.889 -0.3548
15 C1' CT B 14 12 11 1.391 107.688 128.804 0.0394
16 H10 H2 E 15 14 12 1.059 109.468 91.228 0.2007
17 N9 N* S 15 14 12 1.552 105.091 -143.300 -0.0251
18 C8 CK B 17 15 14 1.388 123.797 33.434 0.2006
19 H8 H5 E 18 17 15 1.078 131.047 -3.389 0.1553
20 N7 NB S 18 17 15 1.384 108.967 176.598 -0.6073
21 C5 CB S 20 18 17 1.395 103.021 0.702 0.0515
22 C6 CA B 21 20 18 1.412 128.288 178.252 0.7009
23 N6 N2 B 22 21 20 1.333 125.907 -3.791 -0.9019
24 H60 H E 23 22 21 1.010 120.010 2.829 0.4115
25 H61 H E 23 22 21 1.009 120.014 -177.167 0.4115
26 N1 NC S 22 21 20 1.343 113.319 178.851 -0.7615
27 C2 CQ B 26 22 21 1.340 123.433 -1.664 0.5875
28 H2 H5 E 27 26 22 1.077 120.022 -177.875 0.0473
29 N3 NC S 27 26 22 1.377 125.379 2.123 -0.6997
30 C4 CB E 29 27 26 1.300 108.242 -0.035 0.3053
31 C3' CT M 12 11 10 1.484 117.051 57.581 0.2022
32 H30 H1 E 31 12 11 1.059 117.939 24.820 0.0615
33 O3' OH S 31 12 11 1.407 108.509 149.908 -0.6541
34 H3' HO E 33 31 12 0.967 109.470 163.254 0.4376
35 C2' CT M 31 12 11 1.607 102.425 -96.715 0.0670
36 H20 H1 E 35 31 12 1.059 115.086 78.119 0.0972
37 O2' OH S 35 31 12 1.398 114.943 -153.294 -0.6139
38 H2' HO E 37 35 31 0.967 109.456 -41.679 0.4186
IMPROPER
C8 C4 N9 C1'
C6 H60 N6 H61
N7 N9 C8 H80
N1 N3 C2 H2
C5 N1 C6 N6
LOOP CLOSING EXPLICIT
C1' C2'
C4 C5
C4 N9
DONE
STOPfrcmod.phos ファイルを修正
frcmod.phosファイルをコピーしてAMP.frcmodをつくります- このファイルの
DIHEの部分に下記のパラメータを追加します
O3-P -OS-CT 3 0.375 0.0 3.0- 上記の修正の結果、AMP 用に書き換えたファイル(
AMP.frcmod)は下記のようになります
AMP.frcmod
Remark line goes here
MASS
BOND
ANGLE
ne-c2-o 79.144 117.755 Calculated using o-c2-n2, penalty score= 2.8
ne-ce-ss 64.700 117.230 same as n2-ce-ss, penalty score= 2.8
cd-ce-ss 61.500 117.520 same as ca-ce-ss, penalty score= 2.0
cd-ce-ne 68.300 122.390 same as cc-ce-nf, penalty score= 1.5
ce-cd-ss 61.300 121.580 same as cf-cd-ss, penalty score= 0.8
DIHE
ne-ce-ss-c3 2 2.200 180.000 2.000 same as X -c2-ss-X , penalty score=237.0
cd-ce-ss-c3 1 1.100 180.000 -2.000 same as c2-c2-ss-c3
cd-ce-ss-c3 1 0.700 180.000 3.000 same as c2-c2-ss-c3, penalty score=324.0
nd-cd-ce-ss 4 26.600 180.000 2.000 same as X -ce-cf-X , penalty score=136.0
ss-cd-ce-ss 4 26.600 180.000 2.000 same as X -ce-cf-X , penalty score=136.0
nd-cd-ce-ne 4 26.600 180.000 2.000 same as X -ce-cf-X , penalty score=136.0
ss-cd-ce-ne 4 26.600 180.000 2.000 same as X -ce-cf-X , penalty score=136.0
ce-cd-ss-cc 2 2.200 180.000 2.000 same as X -c2-ss-X , penalty score=232.0
cc-cc-ss-cd 2 2.200 180.000 2.000 same as X -c2-ss-X , penalty score=232.0
cd-cc-ss-cd 2 2.200 180.000 2.000 same as X -c2-ss-X , penalty score=232.0
nd-cd-ss-cc 2 2.200 180.000 2.000 same as X -c2-ss-X , penalty score=232.0
IMPROPER
c3-ne-c2-o 1.1 180.0 2.0 Using the default value
cd-ne-ce-ss 1.1 180.0 2.0 Using the default value
ce-nd-cd-ss 1.1 180.0 2.0 Using the default value
cc-cc-cc-nd 1.1 180.0 2.0 Using the default value
cc-cd-cc-ha 1.1 180.0 2.0 Same as X -X -ca-ha, penalty score= 38.9 (use general term))
c -cc-cd-ha 1.1 180.0 2.0 Same as X -X -ca-ha, penalty score= 38.9 (use general term))
cc-cd-cc-ss 1.1 180.0 2.0 Using the default value
cd-cd-c -o 10.5 180.0 2.0 Using general improper torsional angle X- X- c- o, penalty score= 6.0)
NONBONLcLuc の構造ファイル
- 次のスクリプトを実行すると、LUC の構造データを PDB から取得、基質アナログ (DLSA) を分割して、LUC+OLU+AMP 複合体の構造データ(
LUC_com.pdb)をつくります
prepare.sh
#!/bin/bash
# 構造データをダウンロード
wget https://files.rcsb.org/download/2D1S.pdb
# CSO を CYS に変換
sed -e '/OD CSO/d' -e 's/CSO/CYS/g' 2D1S.pdb > LUC.pdb
# 重複する座標データを削除
awk '{ if (substr($0, 17, 1) == "A") $0 = substr($0, 1, 16) " " substr($0, 18); if (substr($0, 17, 1) != "B") print }' LUC.pdb > LUC_mod.pdb
mv LUC_mod.pdb LUC.pdb
# HETATM ラベルを修正
sed -i -e 's/HETATM/ATOM /g' LUC.pdb
# 発光基質アナログを分割 (split.py が必要です)
python split.py LUC.pdb LUC_com.pdbsh prepare.sh上記を実行するためには、split.pyが必要です。この Python スクリプトは、下記の「詳細」にコードを記載しているので、コピーして使ってください。
- Nakatsu (2006) で報告された LcLuc の構造データ(PDB ID: 2D1S )を PDB からダウンロードします
wget https://files.rcsb.org/download/2D1S.pdb非標準アミノ酸への対処
- この構造では、システイン (
CYS) の一部が、システインスルフォキシド (CSO) という誘導体に置き換わっています
HETATM 441 N CSO A 64 -5.038 4.327 14.691 1.00 5.93 N
HETATM 442 CA CSO A 64 -6.079 4.331 15.718 1.00 6.66 C
HETATM 443 CB CSO A 64 -5.703 3.407 16.877 1.00 6.26 C
HETATM 444 SG CSO A 64 -5.778 1.656 16.397 1.00 10.68 S
HETATM 445 C CSO A 64 -6.358 5.751 16.231 1.00 6.18 C
HETATM 446 O CSO A 64 -7.509 6.116 16.448 1.00 5.96 O
HETATM 447 OD CSO A 64 -7.057 0.904 17.420 1.00 14.68 O CSOなどの非標準アミノ酸は、通常、Amber では取り扱うことができないので、CYSに置き換えますsedコマンドを使って、OD CSOが含まれる行を取り除きますCSOをCYSに置き換えます
sed -e '/OD CSO/d' -e 's/CSO/CYS/g' 2D1S.pdb > LUC.pdbMacOS の場合、sed が標準的なものと異なるので注意 → 詳しくは
こちら
HETATM 441 N CYS A 64 -5.038 4.327 14.691 1.00 5.93 N
HETATM 442 CA CYS A 64 -6.079 4.331 15.718 1.00 6.66 C
HETATM 443 CB CYS A 64 -5.703 3.407 16.877 1.00 6.26 C
HETATM 444 SG CYS A 64 -5.778 1.656 16.397 1.00 10.68 S
HETATM 445 C CYS A 64 -6.358 5.751 16.231 1.00 6.18 C
HETATM 446 O CYS A 64 -7.509 6.116 16.448 1.00 5.96 O 重複する座標データへの対処
- この構造では、一部で、1つの原子に対して複数の座標データが含まれています
ATOM 1271 N ARG A 172 -11.352 5.489 24.197 1.00 9.45 N
ATOM 1272 CA ARG A 172 -12.641 6.147 24.412 1.00 10.12 C
ATOM 1273 C ARG A 172 -13.246 6.695 23.130 1.00 9.90 C
ATOM 1274 O ARG A 172 -14.412 7.101 23.123 1.00 11.23 O
ATOM 1275 CB ARG A 172 -12.511 7.270 25.436 1.00 11.01 C
ATOM 1276 CG ARG A 172 -12.264 6.787 26.866 1.00 12.83 C
ATOM 1277 CD AARG A 172 -13.442 6.086 27.538 0.50 14.51 C
ATOM 1278 CD BARG A 172 -12.363 7.890 27.900 0.50 13.47 C
ATOM 1279 NE AARG A 172 -14.642 6.922 27.575 0.50 16.22 N
ATOM 1280 NE BARG A 172 -11.988 7.426 29.233 0.50 14.80 N
ATOM 1281 CZ AARG A 172 -14.981 7.734 28.573 0.50 15.77 C
ATOM 1282 CZ BARG A 172 -11.891 8.211 30.298 0.50 14.69 C
ATOM 1283 NH1AARG A 172 -14.217 7.845 29.654 0.50 17.25 N
ATOM 1284 NH1BARG A 172 -12.151 9.508 30.202 0.50 15.85 N
ATOM 1285 NH2AARG A 172 -16.098 8.444 28.486 0.50 15.58 N
ATOM 1286 NH2BARG A 172 -11.544 7.695 31.471 0.50 16.45 N - 今回の場合、最初のデータ(
Aとラベルされた方)を使うことにします- PDBデータのなかで、17列目に
Bという文字がある行を削除します - また、17列目に
Aという文字がある場合には空白で置き換えます
- PDBデータのなかで、17列目に
awk '{ if (substr($0, 17, 1) == "A") $0 = substr($0, 1, 16) " " substr($0, 18); if (substr($0, 17, 1) != "B") print }' LUC.pdb > LUC_mod.pdb; mv LUC_mod.pdb LUC.pdbATOM 1271 N ARG A 172 -11.352 5.489 24.197 1.00 9.45 N
ATOM 1272 CA ARG A 172 -12.641 6.147 24.412 1.00 10.12 C
ATOM 1273 C ARG A 172 -13.246 6.695 23.130 1.00 9.90 C
ATOM 1274 O ARG A 172 -14.412 7.101 23.123 1.00 11.23 O
ATOM 1275 CB ARG A 172 -12.511 7.270 25.436 1.00 11.01 C
ATOM 1276 CG ARG A 172 -12.264 6.787 26.866 1.00 12.83 C
ATOM 1277 CD ARG A 172 -13.442 6.086 27.538 0.50 14.51 C
ATOM 1279 NE ARG A 172 -14.642 6.922 27.575 0.50 16.22 N
ATOM 1281 CZ ARG A 172 -14.981 7.734 28.573 0.50 15.77 C
ATOM 1283 NH1 ARG A 172 -14.217 7.845 29.654 0.50 17.25 N
ATOM 1285 NH2 ARG A 172 -16.098 8.444 28.486 0.50 15.58 N HETATM ラベルを修正
- 通常のアミノ酸配列に含まれていない異種原子 (hetero atoms)、つまり、非標準アミノ酸・リガンド・金属イオン・水分子などは、最初の列が
HETATMというタグになっていて、アミノ酸配列のタグ (ATOM) と異なっています
ATOM 4121 N VAL A 545 32.023 44.586 34.701 1.00 30.51 N
ATOM 4122 CA VAL A 545 32.597 43.375 35.295 1.00 31.27 C
ATOM 4123 C VAL A 545 32.494 43.390 36.819 1.00 31.57 C
ATOM 4124 O VAL A 545 32.074 42.404 37.431 1.00 32.25 O
ATOM 4125 CB VAL A 545 34.081 43.165 34.898 1.00 31.34 C
ATOM 4126 CG1 VAL A 545 34.467 41.695 35.044 1.00 31.87 C
ATOM 4127 CG2 VAL A 545 34.352 43.657 33.474 1.00 31.71 C
(省略)
HETATM 4171 O HOH A2002 14.687 19.244 8.432 1.00 5.02 O
HETATM 4172 O HOH A2003 7.144 24.114 15.723 1.00 6.14 O
HETATM 4173 O HOH A2004 15.076 18.773 2.287 1.00 5.55 O - AmberTools や他のツールで扱うときに、
HETATMの部分が連続した配列として扱われなかったことがあるので、念のため、ATOMに置換しておきます
sed -i -e 's/HETATM/ATOM /g' LUC.pdb発光基質アナログを分割
- この構造では、OLU + AMP ではなく、この2つを組み合わせたような構造をもつ DLSA という発光基質の類似体(残基コードは
SLU)が LcLuc に結合しています LUC.pdbから、SLUの部分を抽出してみましょう
grep "ATOM .* SLU " LUC.pdb > SLU.pdbSLU.pdb
ATOM 4131 O39 SLU A2001 17.325 26.177 11.450 1.00 5.69 O
ATOM 4132 C16 SLU A2001 16.264 25.981 10.856 1.00 4.26 C
ATOM 4133 C14 SLU A2001 16.096 24.710 10.113 1.00 3.73 C
(省略)
ATOM 4168 N38 SLU A2001 23.874 24.780 8.175 1.00 6.31 N
ATOM 4169 N37 SLU A2001 21.154 25.902 9.121 1.00 4.97 N
ATOM 4170 C36 SLU A2001 20.318 26.822 9.629 1.00 5.43 C - この
SLUをOLUとAMPに分割します- 手作業でもできますが、ちょっと大変なので、Python スクリプト(
split.py)をつくりました- 1つ目の引数に元のファイル名(
LUC.pdb)、2つ目の引数に出力するファイル名(LUC_com.pdb)を指定します - ここで
_comは、複合体(complex)の略です
- 1つ目の引数に元のファイル名(
- 手作業でもできますが、ちょっと大変なので、Python スクリプト(
python split.py LUC.pdb LUC_com.pdbsplit.py スクリプト
split.py
#!/usr/bin/env python
import sys
input_filename = sys.argv[1]
output_filename = sys.argv[2]
mapping = {
1: ["DELETE"],
2: ["OLU", 1],
3: ["OLU", 2],
4: ["OLU", 3],
5: ["OLU", 6],
6: ["OLU", 5],
7: ["OLU", 4],
8: ["OLU", 7],
9: ["OLU", 8],
10: ["OLU", 9],
11: ["OLU", 10],
12: ["OLU", 15],
13: ["OLU", 16],
14: ["OLU", 11],
15: ["OLU", 12],
16: ["OLU", 13],
17: ["OLU", 14],
18: ["AMP", 2],
19: ["AMP", 1],
20: ["AMP", 3],
21: ["AMP", 4],
22: ["AMP", 5],
23: ["AMP", 6],
24: ["AMP", 7],
25: ["AMP", 9],
26: ["AMP", 10],
27: ["AMP", 11],
28: ["AMP", 12],
29: ["AMP", 13],
30: ["AMP", 8],
31: ["AMP", 14],
32: ["AMP", 23],
33: ["AMP", 22],
34: ["AMP", 21],
35: ["AMP", 20],
36: ["AMP", 17],
37: ["AMP", 18],
38: ["AMP", 19],
39: ["AMP", 16],
40: ["AMP", 15]
}
atomname = {
"OLU": {1: "O6'", 2: "C4'", 3: "N3'", 4: "C2'", 5: "S1'", 6: "C5'", 7: "C2 ", 8: "S1 ", 9: "C5 ", 10: "C6 ", 11: "C4 ", 12: "N3 ", 13: "C9 ", 14: "C8 ", 15: "C7 ", 16: "O7 "},
"AMP": {1: "P ", 2: "O1P", 3: "O2P", 4: "O3P", 5: "O5'", 6: "C5'", 7: "C4'", 8: "O4'", 9: "C3'", 10: "O3'", 11: "C2'", 12: "O2'", 13: "C1'", 14: "N9 ", 15: "C8 ", 16: "N7 ", 17: "C5 ", 18: "C6 ", 19: "N6 ", 20: "N1 ", 21: "C2 ", 22: "N3 ", 23: "C4 "}
}
def read_pdb_file(file_name):
with open(file_name, 'r') as file:
pdb_data = file.readlines()
return pdb_data
def write_pdb_file(file_name, pdb_data):
with open(file_name, 'w') as file:
file.writelines(pdb_data)
def edit_pdb_data(pdb_data, mapping):
edited_pdb_data = []
atom_counter = 1
for line in pdb_data:
if line.startswith('ATOM'):
residue_name = line[17:20].strip()
if residue_name == 'SLU':
residue = mapping[atom_counter][0]
resinum = '2001' if residue != 'AMP' else '2002'
if residue != 'DELETE':
atomnum = mapping[atom_counter][1]
line = line[:13] + atomname[residue][atomnum] + ' ' + residue + ' ' + resinum + line[26:66] + '\n'
else:
line = ''
atom_counter += 1
if line != '':
edited_pdb_data.append(line)
return edited_pdb_data
def main():
pdb_data = read_pdb_file(input_filename)
edited_pdb_data = edit_pdb_data(pdb_data, mapping)
write_pdb_file(output_filename, edited_pdb_data)
if __name__ == "__main__":
main()スクリプトの作り方
- DLSA と OLU および AMP の構造を比較してみます
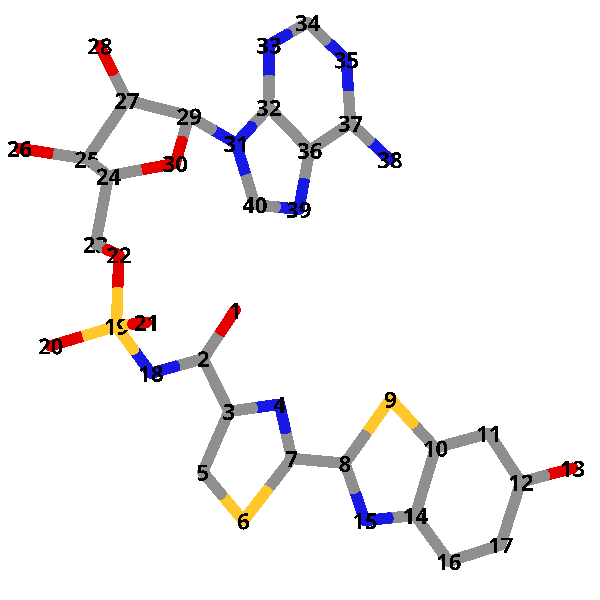
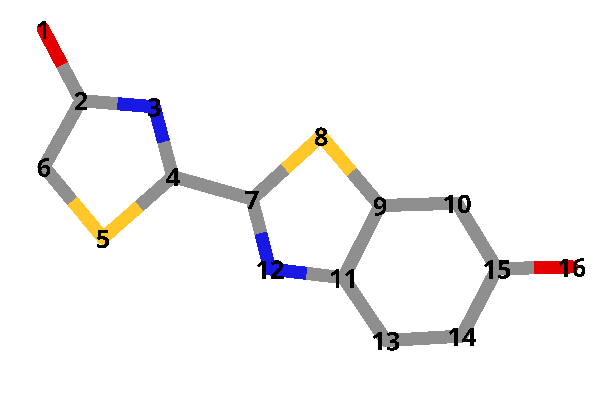
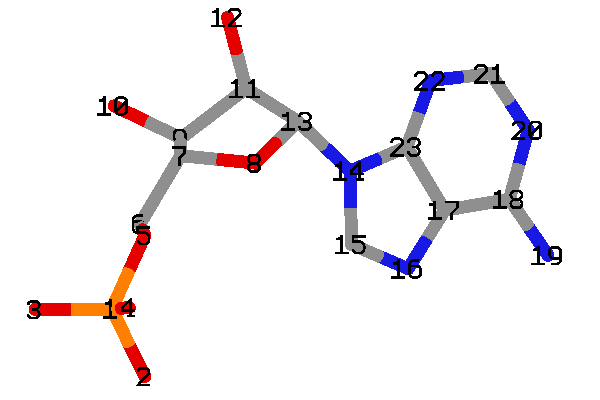
- DLSA の各原子に対して、OLU および AMP が次のように対応していることが分かります
mapping = {
1: ["DELETE"],
2: ["OLU", 1],
3: ["OLU", 2],
4: ["OLU", 3],
5: ["OLU", 6],
6: ["OLU", 5],
7: ["OLU", 4],
8: ["OLU", 7],
9: ["OLU", 8],
10: ["OLU", 9],
11: ["OLU", 10],
12: ["OLU", 15],
13: ["OLU", 16],
14: ["OLU", 11],
15: ["OLU", 12],
16: ["OLU", 13],
17: ["OLU", 14],
18: ["AMP", 2],
19: ["AMP", 1],
20: ["AMP", 3],
21: ["AMP", 4],
22: ["AMP", 5],
23: ["AMP", 6],
24: ["AMP", 7],
25: ["AMP", 9],
26: ["AMP", 10],
27: ["AMP", 11],
28: ["AMP", 12],
29: ["AMP", 13],
30: ["AMP", 8],
31: ["AMP", 14],
32: ["AMP", 23],
33: ["AMP", 22],
34: ["AMP", 21],
35: ["AMP", 20],
36: ["AMP", 17],
37: ["AMP", 18],
38: ["AMP", 19],
39: ["AMP", 16],
40: ["AMP", 15]
}- 上記の対応に基づいて、
SLUをOLUとAMPに分割して、原子名や残基名などを適切に修正する Python スクリプトsplit.pyを作成しました - 対応表を適切に修正すれば、DLASA など、他の発光基質アナログにも対応できます
上記のスクリプトを実行すると、ATOM レコード以外の行を削除したファイルを生成します
上記のスクリプトは「その場しのぎ(ad hoc)」なものなので、適宜修正が必要かもしれません。また、修正すれば、他の発光基質アナログにも対応できるはずです。
OLU+AMP 構造を確認
LUC_com.pdb の OLU+AMP
ATOM 4132 O6' OLU 2001 16.264 25.981 10.856 1.00 4.26
ATOM 4133 C4' OLU 2001 16.096 24.710 10.113 1.00 3.73
ATOM 4134 N3' OLU 2001 17.122 23.880 9.859 1.00 4.69
(省略)
ATOM 4145 N3 OLU 2001 17.161 20.597 8.283 1.00 5.44
ATOM 4146 C9 OLU 2001 18.136 18.459 7.448 1.00 6.18
ATOM 4147 C8 OLU 2001 19.313 17.741 7.265 1.00 6.42
TER
ATOM 4148 O1P AMP 2002 15.163 26.714 10.942 1.00 4.27
ATOM 4149 P AMP 2002 15.068 28.075 11.751 1.00 4.46
ATOM 4150 O2P AMP 2002 13.983 28.834 11.171 1.00 5.13
(省略)
ATOM 4168 N6 AMP 2002 23.874 24.780 8.175 1.00 6.31
ATOM 4169 N7 AMP 2002 21.154 25.902 9.121 1.00 4.97
ATOM 4170 C8 AMP 2002 20.318 26.822 9.629 1.00 5.43
TERLcLuc 複合体の立体構造
- VMD など、使い慣れた分子表示アプリを使って、LcLuc + OLU + AMP 複合体の構造を確認してみましょう
OLU と AMP の酸素原子間の距離が近いので、この図では2つが結合しているように見えますが、構造データ上では異なる分子として分割しています
LuLuc にプロトンを付加する
- PDB からダウンロードした構造には水素が付加されていないため、H+(プロトン)を付加する必要があります
- プロトンの付加は、
PDB2PQR
という Web サービスを使っておこないます
- 論文を書くときには、 関連論文 の引用を忘れないように
準備
- 複合体の構造データ
LUC_com.pdbから、標準アミノ酸&水分子の他(CL,OLU,AMP)を除いた構造データLUC_apo.pdbを作成します
grep -v "ATOM .*AMP " LUC_com.pdb | grep -v "ATOM .*OLU " | grep -v "ATOM .* CL " > LUC_apo.pdbPDB データに含まれる水分子を除去するかどうかには検討の余地がありますが、ここでは Lindh (2010) と同じモデルをつくりたいので、残しておきます
PDB2PQR
- PDB2PQR
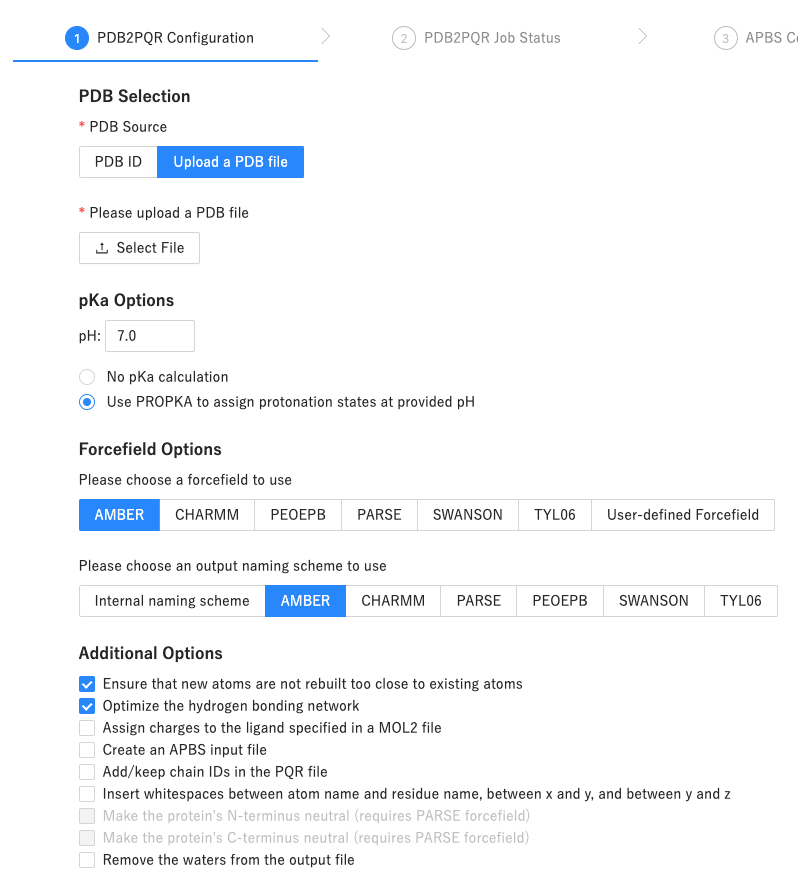
にアクセスして、
LUC_apo.pdbをアップロードします - 各項目を次のような感じに設定したあとで、
Start Jobボタンをクリック
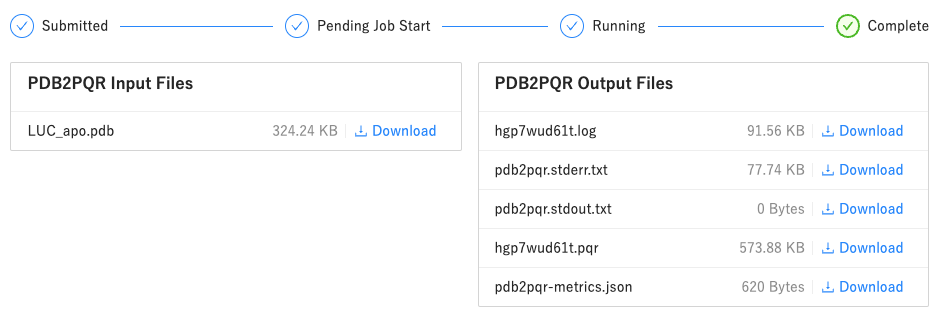
- 作成された
***.pqrファイルをダウンロードします- PQR 形式 は、PDB データに部分電荷と原子半径のパラメータを追加したものです
- 各アミノ酸残基と水分子に、水素(プロトン)が付加されていることを確認してください
LUC_apo.pqr
ATOM 1 N ASP 7 19.436 34.485 -16.440 0.0782 1.8240
ATOM 2 CA ASP 7 20.420 34.912 -15.425 0.0292 1.9080
ATOM 3 C ASP 7 21.266 33.741 -14.951 0.5621 1.9080
ATOM 4 O ASP 7 20.820 32.875 -14.172 -0.5889 1.6612
ATOM 5 CB ASP 7 19.715 35.566 -14.240 -0.0235 1.9080
ATOM 6 CG ASP 7 20.679 36.217 -13.273 0.8194 1.9080
ATOM 7 OD1 ASP 7 21.918 36.157 -13.488 -0.8084 1.6612
ATOM 8 OD2 ASP 7 20.262 36.805 -12.255 -0.8084 1.6612
ATOM 9 H1 ASP 7 19.815 33.732 -16.977 0.2200 0.6000
ATOM 10 HA ASP 7 21.037 35.566 -15.877 0.1141 1.1000
ATOM 11 HB2 ASP 7 19.098 36.261 -14.594 -0.0169 1.4870
ATOM 12 HB3 ASP 7 19.204 34.862 -13.759 -0.0169 1.4870
ATOM 13 H2 ASP 7 18.599 34.181 -15.987 0.2200 0.6000
ATOM 14 H3 ASP 7 19.226 35.252 -17.045 0.2200 0.6000
ATOM 15 N GLU 8 22.516 33.739 -15.400 -0.5163 1.8240
ATOM 16 CA GLU 8 23.451 32.682 -15.063 0.0397 1.9080
(省略)- 下記のアミノ酸残基は複数の異なるプロトン化状態(deprotonated と protonated)があるので、注意が必要です
- 出力された PQR ファイルに含まれていないか、確認しておきましょう
| Deprotonated | Protonated |
|---|---|
| ASP (–) | ASH |
| GLU (–) | GLH |
| HID / HIE | HIP (+) |
grep -E " ASH | GLH | HIP " LUC_apo.pqr- ヒスチジン(
HIS)はプロトンの位置がHID(δ位)とHIE(ε位)で異なるので、こちらも確認しておきましょう
grep -E " HID | HIE " LUC_apo.pqrリガンドを戻す
- プロトンを付加する前に除去したリガンド(
OLUとAMP)の構造データを上記のファイルに追記します- この構造には塩素(
CL)も含まれているのですが、ここでは無視することにします
- この構造には塩素(
grep -v "HETATM.*WAT " LUC_apo.pqr | grep -v "END" > LUC_done.pdb; \
grep "ATOM .*AMP " LUC_com.pdb >> LUC_done.pdb; \
echo "TER" >> LUC_done.pdb; \
grep "ATOM .*OLU " LUC_com.pdb >> LUC_done.pdb; \
echo "TER" >> LUC_done.pdb; \
grep "HETATM.*WAT " LUC_apo.pqr >> LUC_done.pdb; \
echo "TER" >> LUC_done.pdb; \
echo "END" >> LUC_done.pdb塩素(CL)を含めるかどうかは、丁寧に検討する必要がありそうです
Amber で水和構造の準備
- このステップでは、量子化学計算に取り組む前に、 AmberTools を使って、LcLuc + OLU + AMP 複合体の周囲に水分子を配置します
- また、分子力場モデルを用いて、系全体の構造を最適化します
準備
- AmberTools を実行するディレクトリに移動して、
srcというディレクトリを作成します
mkdir src; cd src- 前のステップで準備した LUC+OLU+AMP 複合体構造
LUC_done.pdbをこのsrcディレクトリにコピーします
cp /path/to/LUC_done.pdb ./path/to の部分は、LUC_done.pdb があるディレクトリに置き換えてください。あるいは、sftp などでファイルをアップロードする場合があるかもしれません。
- OLU と AMP の分子力場ファイル(
prepとfrcmod)をダウンロードします
wget https://yamnor.me/a/OLU.tar.gzwget https://yamnor.me/a/AMP.tar.gzOLU.tar.gezとAMP.tar.gzを展開します
tar -zxvf OLU.tar.gztar -zxvf AMP.tar.gz- 展開したファイルを確認すると、
prepとfrcmodファイルがあるはずです
lsAMP.frcmod AMP.prep LUC_done.pdb OLU.frcmod OLU.prep分子研スパコンを使う場合
- 次のコマンドを実行して、AmberTools の環境設定を読み込みます
module load amber水を配置する
- AmberTools の
tleapコマンドを使って、複合体構造(LUC_done.pdb)の周りに水分子を配置した後、Amber を実行するために必要なファイルを生成します
tleap コマンド
tleapコマンドを使って、複合体構造から 5 Å 以内の範囲に、八面体ボックスで、水分子を配置します- 通常の MD シミュレーションでは、10 Å 以内に水分子を配置することが一般的ですが、ここでは QM/MM の準備なので、短い 5 Å に設定しています
- LUC の力場は Amber FF14SB、水分子は TIP3P、リガンドは GAFF を使います
- 系を中性にするために、Na+ と Cl- を自動で追加します
- 今回の場合、Na+ が1個付加されるはずです
tleapの実行コマンドをまとめたスクリプト(tleap.sh)を準備しました
tleap.sh
#!/bin/bash
cat <<EOF >| tleap.cmd
source leaprc.gaff
source leaprc.protein.ff14SB
source leaprc.water.tip3p
loadAmberParams frcmod.ions234lm_1264_tip3p
loadAmberParams src/OLU.frcmod
loadAmberPrep src/OLU.prep
loadAmberParams src/AMP.frcmod
loadAmberPrep src/AMP.prep
com = loadPDB src/LUC_done.pdb
solvateOCT com TIP3PBOX 5.0
addIons com Na+ 0
addIons com Cl- 0
savePDB com amb.pdb
saveAmberParm com amb.top amb.crd
EOF
tleap -f tleap.cmd
rm tleap.cmdsrcと同じ階層のディレクトリでtleap.shを実行します
sh tleap.sh- 座標ファイル(
amb.crd)とトポロジーファイル(amb.top)が生成されるはずです
構造最適化
準備
1_minというディレクトリを作成しておき、出力ファイルをこの中に保存することにします
mkdir 1_minインプットファイル
- AmberTools の
sanderコマンドを使って、構造最適化をおこなう場合のインプットファイル(1_min.inp)は、次のようになります
1_min.inp
Protein
&cntrl
imin = 1,
ntx = 1,
irest = 0,
ntpr = 100,
ntwr = 100,
ntwx = 100,
ntwprt = 0,
maxcyc = 20000,
ntmin = 0
ntc = 1,
ntf = 1,
ntb = 1,
cut = 8.0,
/ジョブスクリプト
- 分子研スパコンなどで、キューイングシステム PBS (Portable Batch System) を用いて計算を実行する場合は、次のようなジョブスクリプト(
1_min.job)を準備します
1_min.job
#!/bin/bash
#PBS -l select=1:ncpus=8:mpiprocs=8:ompthreads=1:jobtype=core
#PBS -l walltime=1:00:00
MPI=8
OMP=1
JOB=1_min
PRE=amb
INP=../${JOB}.inp
TOP=../amb.top
INI=../amb.crd
AMB="mpirun -n ${MPI} sander.MPI"
module load amber
cd ${PBS_O_WORKDIR}/${JOB}
if [ -f ${INI} ]; then
cp ${INI} ${PRE}.ref
else
echo "Error:1"
exit 1
fi
if [ -f ${INP} -a -f ${TOP} ]; then
${AMB} \
-O \
-i ${INP} \
-p ${TOP} \
-c ${INI} \
-o ${PRE}.out \
-r ${PRE}.crd \
-x ${PRE}.trj \
-ref ${PRE}.ref \
-inf ${PRE}.inf
else
echo "Error:2"
exit 1
fi
if [ -f ${PRE}.crd ]; then
ambpdb -p ${TOP} -c ${PRE}.crd > ${PRE}.pdb
else
echo "Error:3"
exit 1
fiMPI 並列数などを適宜変更してください。冗長な部分が多いので、適宜削除して使ってください。
実行
1_minと1_min.inpと同じディレクトリで、1_min.jobスクリプトを実行します
jsub 1_min.job- ジョブが正常に終了すると、
1_minディレクトリ内に出力ファイルが生成されます
cd 1_min; lsamb.crd amb.inf amb.out amb.pdb amb.ref amb.trj結果の確認
- VMD などの分子表示ソフトを使って、構造最適化後の PDB ファイル(
amb.pdb)で、水和構造を確認してみましょう
- 初期構造では、OLU と AMP が近接していましたが、最適化後にはこれらの分子が離れて、適切な位置になっています
水和構造の抽出
cpptrajコマンドを使って、構造最適化後の座標ファイル(amb.crd)から、複合体の周囲 5Å以内の水分子を含めた水和構造を抽出します- 次のスクリプトを使って、水和構造の座標ファイル(
amb_wat.rst7)と、これに対応するトポロジーファイル(amb_wat.top)を生成します- 確認用に、PDB 形式のファイル(
amb_wat.pdb)も生成しておきましょう
- 確認用に、PDB 形式のファイル(
cpptraj.sh
#!/bin/bash
module load amber
cmd="
parm ../amb.top
trajin amb.crd
reference amb.crd
center :1-541
autoimage familiar
strip :Na+,Cl-
strip !(:1-541<:5.0)
trajout amb_wat.pdb
trajout amb_wat.rst7 restart
"
echo "${cmd}" | cpptraj
cmd="
parm ../amb.top
reference amb.crd
center :1-541
parmstrip :Na+,Cl-
parmstrip !(:1-541<:5.0)
parmwrite out amb_wat.top
"
echo "${cmd}" | cpptraj1_minディレクトリで、上記のスクリプトを実行します
sh cpptraj.sh- VMD などを使って、できた水和構造を確認してみましょう
Gaussian で量子化学計算
- このステップでは、 Gaussian を使って、LcLuc + OLU + AMP 複合体の量子化学計算に取り組みます
- 計算手法としては、ONIOM 法を用いて、OLU のみを量子化学計算の対象として、残りの部分(LUC, AMP, 水分子)は力場モデルで扱います
- はじめに、基底状態で構造最適化をおこなった後、次に、励起状態の解析をおこないます
準備
pdb2oniom とは?
- ONIOM 計算用に Gaussian の入力ファイルを準備するために、今回、
pdb2oniom
という Python スクリプトを使います
- pdb2oniom は、東京医科歯科大の 森脇さん が開発されています
- Gaussian には Amber の力場パラメータ が準備されているので、PDB ファイルを GaussView で読み込めば、このスクリプトを使わなくても ONIOM 計算はできます
- このスクリプトを使うと、Amber で準備したトポロジーファイルから力場パラメータを読み込んで、ONIOM 計算に反映させることができます
- たとえば、Amber で MD をおこなった後で ONIOM 計算をおこなうようなときには、ほぼ同じ力場パラメータを使うので、構造最適化の収束性が良いことが期待できます
- また、MM 領域に非標準アミノ酸を含む場合などは、Gaussian 標準の力場パラメータでは対応できませんが、このスクリプトを使うと便利です
pdb2oniom のインストール
- GitHub のページ
にある説明を参考に、
pdb2oniomをインストールします
python -m pip install git+https://github.com/BILAB/pdb2oniom.gitpdb2oniom_py [-h] -p PARMFILE -r RST7FILE -resid RESID [-n NEAR] [-o OUTPUTFILE] [--improper] [-m MEM] [--nproc NPROC]pdb2oniom のオプション
-h: ヘルプを表示-p PARMFILE: トポロジーファイル(parm7 ファイル)のパスを指定-r RST7FILE: 座標ファイル(rst7 ファイル)のパスを指定-resid RESID: QM 領域に指定する残基のリストが含まれるファイルのパスを指定- このファイルでは、残基の名前とIDを
[残基名] "残基ID"という形式で指定
- このファイルでは、残基の名前とIDを
-n NEAR: QM 領域からNEARÅ 以内にある残基を構造最適化の対象とする-o OUTPUTFILE: Gaussian 入力ファイルのファイル名を指定-m MEM: Gaussian 入力ファイルで指定するメモリ使用値を指定--nproc NPROC: Gaussian 入力ファイルで指定するプロセッサ数を指定
基底状態の構造最適化
準備
- Gaussian を実行するディレクトリに移動して、
a..S0というディレクトリを作成しますS0は、基底状態を意味します
mkdir a..S0; cd a..S0計算手法や計算対象ごとにディレクトリを分けるのは「個人的な好み」ですが、後で見返すときに便利ですよ。
- Amber で生成した水和構造の座標ファイル(
amb_wat.rst7)とトポロジーファイル(amb_wat.top)をこのディレクトリにコピーします
cp /path/to/amb_wat.rst7 /path/to/amb_wat.top ./path/to は、amb_wat.rst7 と amb_wat.top がある場所に置き換えてください
pdb2oniom の実行
- 今回は OLU のみを QM 領域として扱いたいので、
pdb2oniom.qmという名前のファイルに、次のように指定します- このファイルでは、残基の名称 (ResName) と番号 (ResID) を
[ResName] "ResID"で指定します
- このファイルでは、残基の名称 (ResName) と番号 (ResID) を
pdb2oniom.qm
[OLU] "541"- 下記のコマンドを実行して、Gaussian の入力ファイル(
a_S0.com)を生成します- 今回は、QM 領域に指定した OLU と、その周囲 7 Å 以内にあるアミノ酸残基を構造最適化の対象とします
pdb2oniom_py -p amb_wat.top -r amb_wat.rst7 --resid pdb2oniom.qm -n 7 -o opt.gjf2024-05-07 08:29:59,772 pdb2oniom.py:782 - INFO - pdb2oniom.py ver.0.3.2: Amber parm and rst7 files to Gaussian ONIOM input file.
2024-05-07 08:30:00,463 pdb2oniom.py:691 - INFO - Total charge of low layer is calculated -1
2024-05-07 08:30:00,463 pdb2oniom.py:692 - INFO - Core residues list file pdb2oniom.qm provided.
2024-05-07 08:30:00,463 pdb2oniom.py:693 - INFO - All residues within 7.0 Å from core region are free to move (0) during geometry optimization.
2024-05-07 08:30:00,545 pdb2oniom.py:146 - INFO - 758 atoms are set to be movable during geometry optimization.
2024-05-07 08:30:01,204 pdb2oniom.py:705 - INFO - Opening file opt.gjf for output ...
2024-05-07 08:30:01,208 pdb2oniom.py:708 - INFO - Successfully wrote opt.gjf file.
2024-05-07 08:30:01,208 pdb2oniom.py:709 - INFO - Opening file opt.gjf.onb for output ...
2024-05-07 08:30:01,210 pdb2oniom.py:713 - INFO - Successfully wrote opt.gjf.onb file.
2024-05-07 08:30:01,210 pdb2oniom.py:714 - INFO - pdb2oniom.py ends.
2024-05-07 08:30:01,210 pdb2oniom.py:715 - INFO - NOTE: The charges and multiplicity of high layer should be modified for your calculations. Please use GaussView 6 to modify the QM regions and link atoms.- 生成された Gaussian の入力ファイル(
opt.gjf)を確認してみます- 各原子ごとに、atomtype、部分電荷、構造最適化フラグなどが設定されています
- また、QM 領域と MM 領域の原子が正しく分類されています
- 力場パラメータとして、Amber で準備したトポロジーファイルから読み込まれています
opt.gjf
%chk=opt.chk
%mem=60GB
%nprocshared=16
#p opt=(quadmac,calcfc,maxstep=5) oniom(b3lyp/6-31g(d,p):amber=softonly)
scf=(xqc,intrep,maxconventionalcyc=80) geom=connectivity nosymm iop(2/15=3) Amber=(FirstEquiv)
ONIOM inputfile generated by pdb2oniom.py from amb_wat.top and amb_wat.rst7.
Please use GaussView 6 to modify the qm region and add the link atom info.
-1 1 0 1 0 1
N-N3-0.078200 -1 68.187 60.503 45.161 L
H-H-0.220000 -1 69.050 60.619 44.639 L
H-H-0.220000 -1 67.471 60.208 44.504 L
(省略)
H-H1-0.097200 0 49.345 44.314 49.117 L
O-OH--0.613900 0 48.410 43.979 50.944 L
H-HO-0.418600 0 48.905 44.720 51.374 L
O-LO--0.563608 0 44.798 43.521 45.596 H
C-LC2-0.616170 0 45.045 44.011 44.499 H
C-LC3-0.028279 0 44.459 45.312 43.991 H
(省略)
13801
13802 13801 1.0
13803 13801 1.0 13802 1.0
HrmStr1 C CA 469.0 1.409
HrmStr1 C N 490.0 1.335
HrmStr1 C O 570.0 1.229
(省略)インプットの修正
- ここで、Gaussian の計算を実行する前に、多くの場合、入力ファイルの次の部分を修正する必要があります
- QM 領域の部分電荷とスピン多重度:
- 今回の場合、OLU の電荷は
-1、スピン多重度は1です- 該当部分を
-1 1 -1 1 -1 1に修正します
- 該当部分を
- 今回の場合、OLU の電荷は
- 計算手法:
- 今回はとりあえず、
B3LYPを汎関数とする DFT 計算をおこないます- 光吸収・発光過程を解析するときには、長距離補正を含む
wB97XDやCAM-B3LYPなどを使うこともあります
- 光吸収・発光過程を解析するときには、長距離補正を含む
- 今回はとりあえず、
- 基底関数:
- 今回はとりあえず、
6-31G(d)を使うことにしました- 励起状態の解析をおこなう場合には、分散関数を加えた
6-31+G(d,p)やaug-cc-pVTZなどを使うこともあります
- 励起状態の解析をおこなう場合には、分散関数を加えた
- 今回はとりあえず、
- QM/MM 境界の Link 原子:
- 今回は、設定する必要はありません
- この情報は、GaussView を使うと簡単に追加することができます
- 今回は、設定する必要はありません
- プロセス数やメモリ:
- 分子研スパコンを使う場合、
g16subコマンドがmemとnprocsharedを自動で設定するので、削除しておきます
- 分子研スパコンを使う場合、
opt.gjf
%chk=opt.chk
#p opt=(quadmac,calcfc,maxstep=5) oniom(b3lyp/6-31g(d):amber=softonly)
scf=(xqc,intrep,maxconventionalcyc=80) geom=connectivity nosymm iop(2/15=3) Amber=(FirstEquiv)
LUC+OLU+AMP / ONIOM / Opt / S0
-1 1 -1 1 -1 1
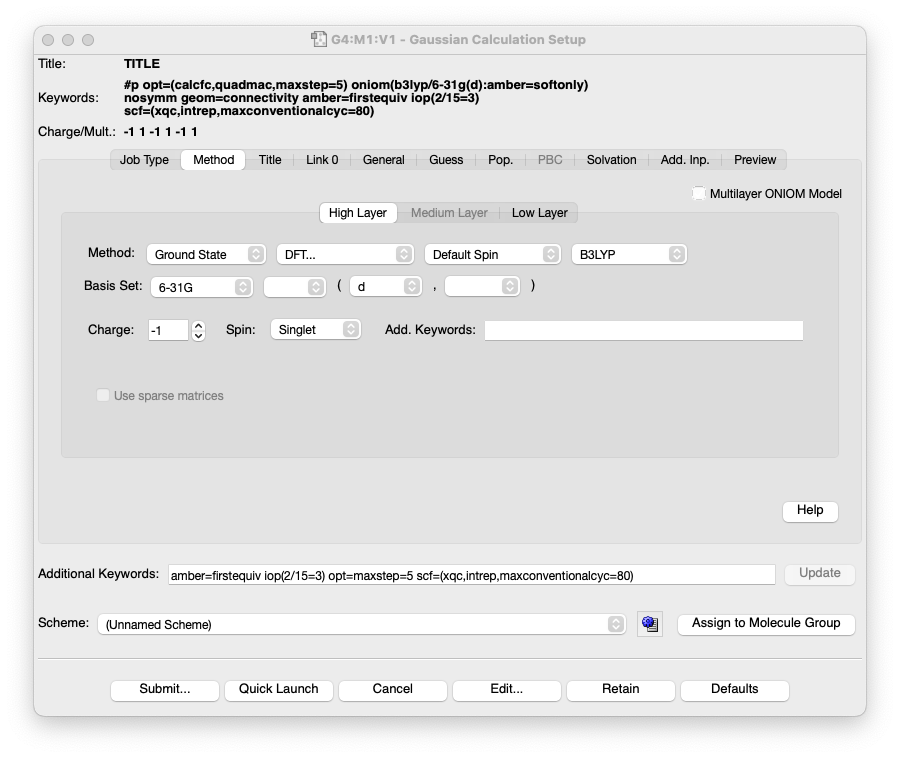
(省略)- Gaussian の入力ファイルを GaussView で読み込んで、確認してみます
量子化学計算の実行
- 準備した Gaussian の入力ファイル(
opt.gjf)を使って、計算を実行します- 分子研スパコンを使う場合は、
g16subコマンドを使って計算を実行します
- 分子研スパコンを使う場合は、
g16sub opt.gjf結果の確認
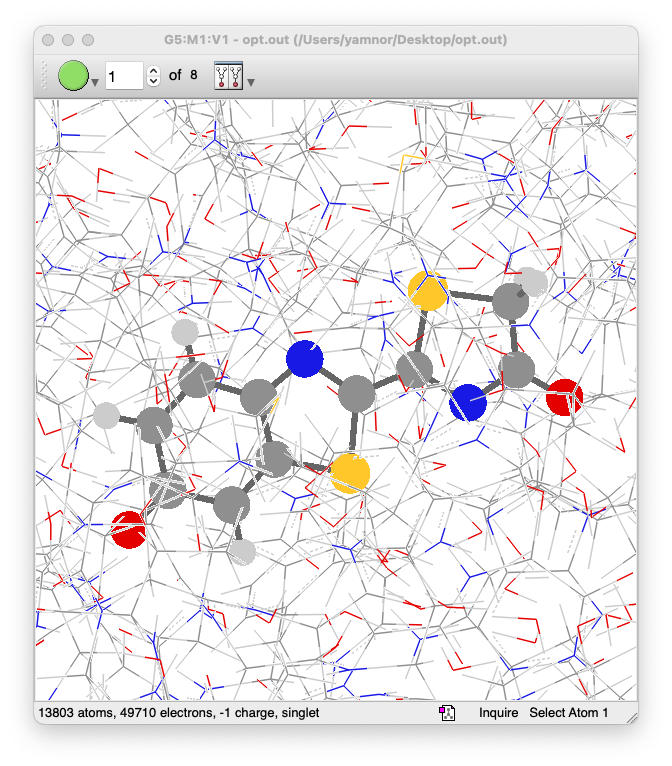
- Gaussian の計算が正常に終了したら、出力ファイル(
opt.out)を GaussView で開いて、構造最適化の結果を確認してみましょう - 今回の場合、OLU とその周囲のアミノ酸残基だけを最適化しているので、この部分を注意深くチェックしましょう
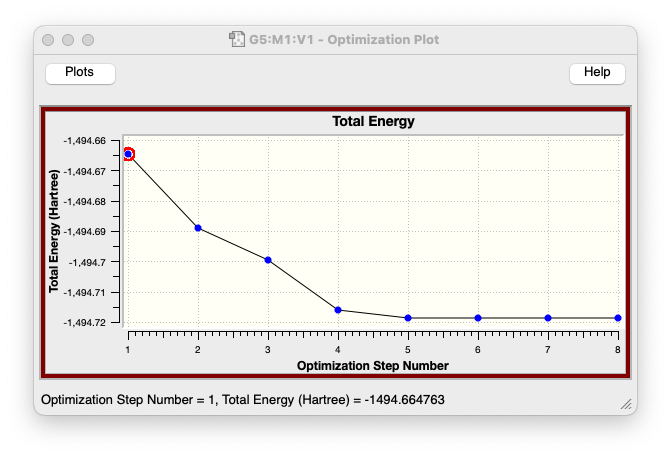
- 構造最適化が収束に向かうことで、ポテンシャルエネルギーの値が段々と小さくなっていく様子も確認できますね
- 無事に基底状態の構造最適化が完了したことを確認できたら、次に、励起状態の解析に取り組みます
励起状態の構造最適化
- 今回は、励起状態の解析をおこなうために、 TD-DFT 計算をおこないます
- 発光過程を解析するためには、基底状態で最適化した構造を出発点として、励起状態での構造最適化もおこなう必要があります
準備
- Gaussian を実行するディレクトリに移動して、
b..S1というディレクトリを作成しますS1は、基底状態を意味します
mkdir b..S1; cd b..S1- 基底状態の構造最適化で用いた入力ファイル(
opt.gjf)、出力されたチェックポイントファイル(opt.chk)をこのディレクトリにコピーします
cp ../a..S0/opt.gjf ../a..S0/opt.chk .- 励起状態の解析をおこなうために、Gaussian の入力ファイル(
opt.gjf)を次のように修正します- 構造データは、チェックポイントファイル(
opt.chk)から読み込むように設定します - TD-DFT を実行するためのオプション(
TD)を追加します
- 構造データは、チェックポイントファイル(
opt.gjf
%chk=opt.chk
#p opt=(quadmac,maxstep=5) oniom(wb97xd/6-31g(d) td:amber=softonly)
scf=(xqc,intrep,maxconventionalcyc=80) geom=connectivity nosymm iop(2/15=3) Amber=(FirstEquiv)
geom=check
LUC+OLU+AMP / ONIOM / Opt / S1
-1 1 -1 1 -1 1
1 2 1.0 3 1.0 4 1.0 5 1.0
2
3
(省略)量子化学計算の実行
- 基底状態での構造最適化と同様に、準備した Gaussian の入力ファイル(
opt.gjf)を使って、計算を実行します- 分子研スパコンを使う場合は、
g16subコマンドを使って計算を実行します
- 分子研スパコンを使う場合は、
g16sub opt.gjf結果の確認
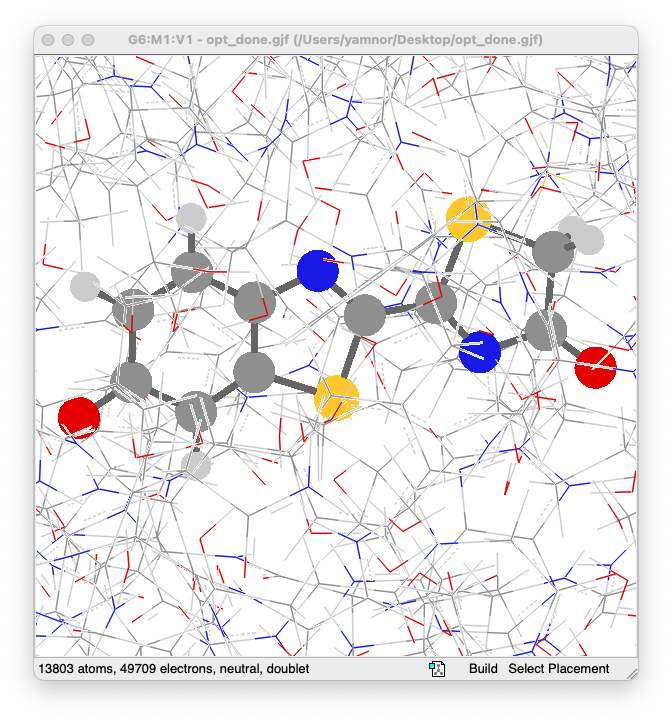
- Gaussian の計算が正常に終了したら、出力ファイル(
opt.out)を GaussView で開いて、構造最適化の結果を確認してみます- 基底状態の最適化構造から、大きくは変化していないように見えますね
- 励起状態の構造最適化をおこなったので、発光波長を確認することができます
- 構造最適化では、励起状態の計算を何度も繰り返しているので、最後の部分を確認します
opt.out
(省略)
Excitation energies and oscillator strengths:
Excited state symmetry could not be determined.
Excited State 1: Singlet-?Sym 2.5613 eV 484.07 nm f=0.6756 <S**2>=0.000
64 -> 65 0.69291
This state for optimization and/or second-order correction.
Total Energy, E(TD-HF/TD-DFT) = -1440.12213510
Copying the excited state density for this state as the 1-particle RhoCI density.
Excited state symmetry could not be determined.
Excited State 2: Singlet-?Sym 3.4412 eV 360.29 nm f=0.0000 <S**2>=0.000
63 -> 65 0.65633
63 -> 67 0.18223
63 -> 70 -0.13934
Excited state symmetry could not be determined.
Excited State 3: Singlet-?Sym 3.7633 eV 329.45 nm f=0.1862 <S**2>=0.000
62 -> 65 0.68324
SavETr: write IOETrn= 770 NScale= 10 NData= 16 NLR=1 NState= 3 LETran= 64.
The selected state is a singlet.
CISGrd: IGrad=2 NXY=2 DFT=T
CISAX will form 1 AO SS matrices at one time.
NMat= 1 NSing= 1 JSym2X= 0.
Leave Link 914 at Sun Feb 25 14:35:24 2024, MaxMem= 2516582400 cpu: 1688.2 elap: 108.2
(Enter /apl/gaussian/16c02/g16/l1002.exe)
(省略)- この結果を見ると、複合体中の OLU の発光波長は 484 nm と推測されることがわかります
- 実は、この場合の計算は、QM 領域として設定した OLU と、MM 領域として設定した他の部分(LUC, AMP, 水分子)の静電的な相互作用を無視して計算しているので、適切な推測ではない可能性があります
- 最後のステップでは、励起状態の最適化構造に基づいて、QM/MM の静電的な相互作用を考慮した計算をおこないます
励起状態の一点計算
準備
- 励起状態の構造最適化計算で用いた入力ファイル(
opt.gjf)とチェックポイントファイル(opt.chk)を使って、一点計算をおこないます
cp opt.gjf sng.gjf; cp opt.chk sng.chk- Gaussian の入力ファイル(
sng.gjf)を次のように修正します- 構造データは、チェックポイントファイル(
sng.chk)から読み込むように設定します - 一点計算をおこなうため、構造最適化のオプション(
opt)を削除します - QM/MM の静電的な相互作用を考慮するため、
EmbedChargeを追記します- MM 領域にある原子上に力場パラメータとして設定した部分電荷を埋め込み、QM 領域の量子化学計算に反映させることで、静電的な相互作用に伴って波動関数の分極を考慮します
- 発光スペクトルの解析範囲を広げるため、
td=(nstates=10)を追記します
- 構造データは、チェックポイントファイル(
sng.gjf
%chk=sng.chk
#p oniom(wb97xd/6-31g(d) td=(nstates=10):amber=softonly)=EmbedCharge geom=check density=current
scf=(xqc,intrep,maxconventionalcyc=80) geom=connectivity nosymm iop(2/15=3) Amber=(FirstEquiv)
TITLE
-1 1 -1 1 -1 1
1 2 1.0 3 1.0 4 1.0 5 1.0
2
3
(省略)量子化学計算の実行
- 準備した Gaussian の入力ファイル(
sng.gjf)を使って、計算を実行します- 分子研スパコンを使う場合は、
g16subコマンドを使って計算を実行します
- 分子研スパコンを使う場合は、
g16sub sng.gjf結果の確認
- 出力ファイル(
sng.out)のなかで、発光波長の値を確認してみます
sng.out
(省略)
Excited state symmetry could not be determined.
Excited State 1: Singlet-?Sym 2.4769 eV 500.57 nm f=0.6143 <S**2>=0.000
62 -> 65 0.11191
64 -> 65 0.69184
This state for optimization and/or second-order correction.
Total Energy, E(TD-HF/TD-DFT) = -2290.10091116
Copying the excited state density for this state as the 1-particle RhoCI density.
Excited state symmetry could not be determined.
Excited State 2: Singlet-?Sym 3.3859 eV 366.18 nm f=0.0000 <S**2>=0.000
63 -> 65 0.66051
63 -> 67 0.16456
63 -> 70 -0.13853
Excited state symmetry could not be determined.
Excited State 3: Singlet-?Sym 3.6712 eV 337.72 nm f=0.2296 <S**2>=0.000
62 -> 65 0.68307
64 -> 65 -0.10426
(省略)- 複合体中の OLU の発光波長は、静電的な相互作用を考慮する前は 484 nm と推測されていましたが、静電的な相互作用を考慮することで長波長側に 16 nm 程度シフトした 501 nm と推測されることがわかります
- もしかすると、シフトの値が意外に小さいので、OLU の発光に対する LUC の環境の影響は小さい?と思うかも知れませんが、私たちが現在取り組んでいる解析の結果から、OLU の発光波長は LUC によって巧みに制御されていることがわかっています


