背景
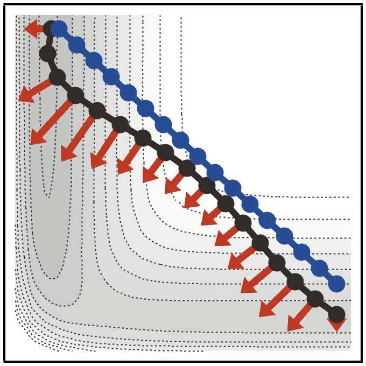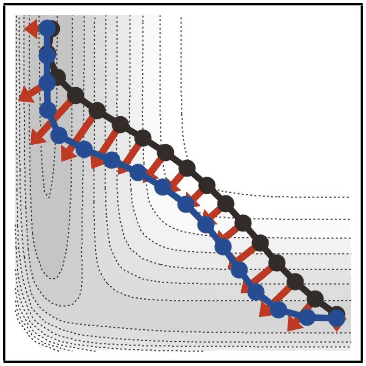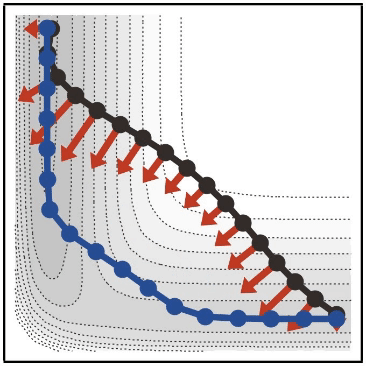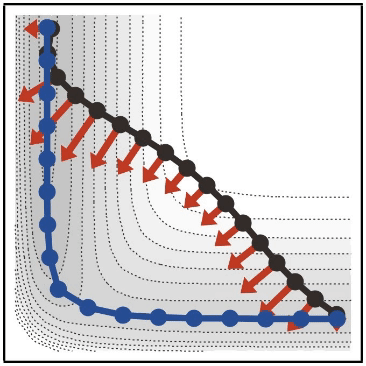
- 最小エネルギー探索は、化学反応などにおいて、始状態(反応系)から終状態(生成系)に辿り着くまでのエネルギー変化が最も低くなるような経路を見つけるための手法です
- QM/MM 自由エネルギー摂動法を用いて自由エネルギー変化を解析しようとするときには、MEP を探索することが必要になります
- string 法は、最小エネルギー探索手法の一つで、E や Vanden-Eijnden らによって開発されました(
論文
)
- 類似の方法としては、Nudged Elastic Band (NEB) 法などがあります(
論文
)
- NEB 法については、 Reaction plus という有料のソフトウェアや、 ASE という無料の Python ライブラリでも利用できます
- 類似の方法としては、Nudged Elastic Band (NEB) 法などがあります(
論文
)
- 山本が string 法について紹介した 日本語のレビュー もあるので、参考にしてみてください
実行
qsub optpath_in.rb- ジョブの実行状況は、
qstatコマンドで確認できます
qstatJob id Name User Time Use S Queue
---------------- ---------------- ---------------- -------- - -----
2893390.fe3-adm ethylene yamnor 00:08:29 R SMALL- 計算が始まると、今回の設定では、
nodeとpathという2つのフォルダ、node.log.*とpath.log.*という2つのファイルができます
ethylene/
├─node/
├─node.log.2893390.fe3-adm
├─optpath_in.rb
├─path/
├─path.log.2893390.fe3-adm
├─path.xyz
├─qm.grad
└─qm.sngnodeには、各ノードでの Q-Chem の計算ログが残ります
node/
├─00
│ ├─qm.geom
│ ├─qm.grad
│ ├─qm.inp
│ └─qm.out
├─01
└─02
(省略)
├─09
├─10
└─11pathには、最適化している経路の情報が記録されます
path/
├─000.dat
├─000.grad
├─000.xyz
├─001.dat
├─001.grad
└─001.xyz
(省略)
├─030.dat
├─030.grad
├─030.xyz
└─sng.xyz*.datには、経路上の各ノードごとのポテンシャルエネルギーを記録します
030.dat
# Energy / au Delta-E / au Gradient / au DE / kcal mol-1
-78.3353613584 0.0000000000 0.0529956323 0.0000000000
-78.3716415974 0.0000000000 0.0005435334 -22.7661946372
-78.3741964515 0.0000000000 0.0003266565 -24.3693898411
-78.3792345117 0.0000000000 0.0004439576 -27.5308204999
-78.3859990766 0.0000000000 0.0008330186 -31.7756492086
-78.3928286549 0.0000000000 0.0016623470 -36.0612744495
-78.3984990789 0.0000000000 0.0005942788 -39.6195194280
-78.4018534843 0.0000000000 0.0019813852 -41.7244406387
-78.4016302569 0.0000000000 0.0021692475 -41.5843633247
-78.3998829011 0.0000000000 0.0021710707 -40.4878810030
-78.3893126226 0.0000000000 0.0011168969 -33.8549308363
-78.3573213260 0.0000000000 0.0179266038 -13.7800882546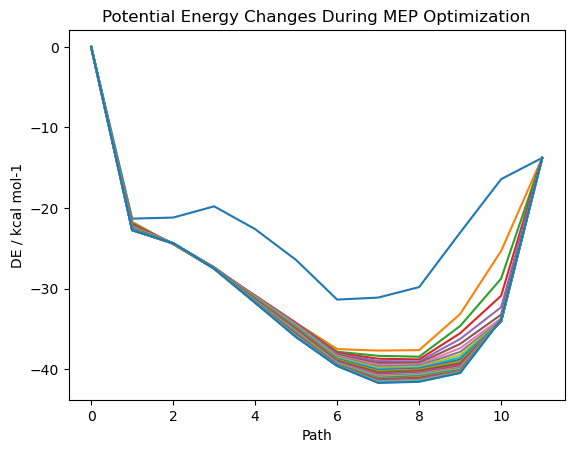
このファイルは、SF-TD 計算の参照状態、つまり、T1 のエネルギー変化が出力されるので、今回はあまり役に立たない情報になってしまっています
import matplotlib.pyplot as plt
import numpy as np
# データファイルの名前のリストを生成
filenames = [f"path/{i:03}.dat" for i in range(31)]
# 各ファイルからデータを読み込み、プロットする
for filename in filenames:
# ファイルからデータを読み込む
data = np.loadtxt(filename, skiprows=1) # ヘッダー行をスキップ
potential_energy_changes = data[:, 3] # 4列目のデータを取得
# データをプロット
plt.plot(potential_energy_changes, label=f"File {filename}")
# グラフの設定
plt.title('Potential Energy Changes During MEP Optimization')
plt.xlabel('Path')
plt.ylabel('DE / kcal mol-1')
plt.show()*.graには、経路上の各ノードで原子に掛かるエネルギー勾配の大きさを記録します
030.gra
6
C -0.2184073660 0.0000176576 -0.0000391570
C 0.2184259137 0.0000059386 0.0000392522
H 0.0001195234 -0.0040814767 -0.0000024149
H 0.0001206082 0.0040754422 0.0000023091
H -0.0001359842 0.0040930123 -0.0000022638
H -0.0001226951 -0.0041105742 0.0000022745
6
C -0.0016749522 -0.0000800634 0.0006689344
C 0.0016567148 -0.0000073016 -0.0002819993
H 0.0018830068 -0.0002684249 -0.0012523324
(省略)*.xyzには、経路上の各ノードにおける分子の座標を記録します
000.xyz
6
C 0.0051800000 0.0000000000 0.0011200000
C -1.3151200000 0.0000100000 0.0008900000
H -1.8839800000 -0.9165800000 0.0008200000
H -1.8839800000 0.9165900000 0.0007900000
H 0.5739400000 0.9166500000 0.0012300000
H 0.5739500000 -0.9166800000 0.0011600000
6
C 0.1069247886 0.0010471256 -0.0151659909
C -1.4163011623 -0.0002422812 0.0087023126
H -1.9487916403 -0.9294334570 0.0335358554
(省略)結果
nodeフォルダにある Q-Chem の出力ファイル(qm.out)かr、SF-TD 法で計算した S0 および S1 状態のエネルギーを読み取ることができます
qm.out
(省略)
---------------------------------------------------
SF-DFT Excitation Energies
(The first "excited" state might be the ground state)
---------------------------------------------------
Excited state 1: excitation energy (eV) = -4.1670
Total energy for state 1: -78.52286339 au
<S**2> : 0.0133
S( 2) --> S( 1) amplitude = 0.9899 alpha
Excited state 2: excitation energy (eV) = 0.9352
Total energy for state 2: -78.33536136 au
<S**2> : 2.0182
S( 1) --> S( 1) amplitude = 0.7107 alpha
S( 2) --> S( 2) amplitude = 0.6939 alpha
Excited state 3: excitation energy (eV) = 4.0106
Total energy for state 3: -78.22234102 au
<S**2> : 1.0134
D( 7) --> S( 1) amplitude = -0.9886
(省略)- 次のような Python スクリプトで、各ノードのエネルギーを読み取り、グラフにしてみます
import os
import matplotlib.pyplot as plt
# eVからkcal/molへの変換係数
ev_to_kcalmol = 23.06035
# フォルダ名を生成
folder_names = [f"node/{i:02}" for i in range(12)]
# エネルギー値を格納するリスト
energies_state1 = []
energies_state2 = []
# 各フォルダのファイルを読み込み
for folder in folder_names:
filepath = os.path.join(folder, "qm.out")
with open(filepath, 'r') as file:
energy_state1 = None
energy_state2 = None
for line in file:
if "Excited state 1: excitation energy (eV) =" in line:
energy_ev = float(line.split('=')[1].strip())
energy_kcalmol = energy_ev * ev_to_kcalmol # eVからkcal/molに変換
energy_state1 = energy_kcalmol
elif "Excited state 2: excitation energy (eV) =" in line:
energy_ev = float(line.split('=')[1].strip())
energy_kcalmol = energy_ev * ev_to_kcalmol # eVからkcal/molに変換
energy_state2 = energy_kcalmol
# 両方のエネルギーが見つかったらループを抜ける
if energy_state1 is not None and energy_state2 is not None:
break
energies_state1.append(energy_state1)
energies_state2.append(energy_state2)
# エネルギーの変化を計算(始点をゼロに設定)
delta_energies_state1 = [e - energies_state2[0] for e in energies_state1]
delta_energies_state2 = [e - energies_state2[0] for e in energies_state2]
# グラフをプロット
#plt.plot(delta_energies_state1, marker='o', label='S0', color='skyblue')
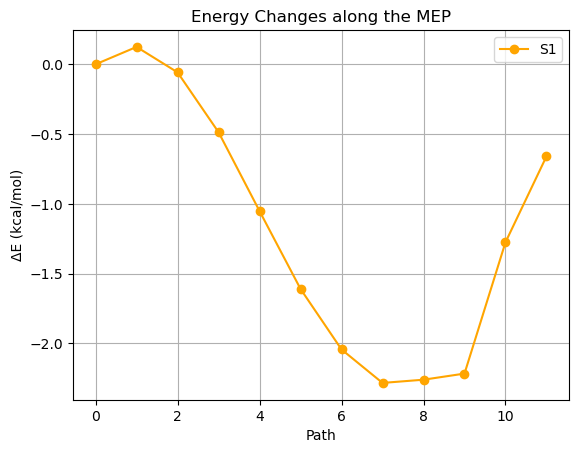
plt.plot(delta_energies_state2, marker='o', label='S1', color='orange')
plt.title('Energy Changes along the MEP')
plt.xlabel('Path')
plt.ylabel('ΔE (kcal/mol)')
#plt.ylabel('ΔE (eV)')
plt.legend()
plt.grid(True)
plt.show()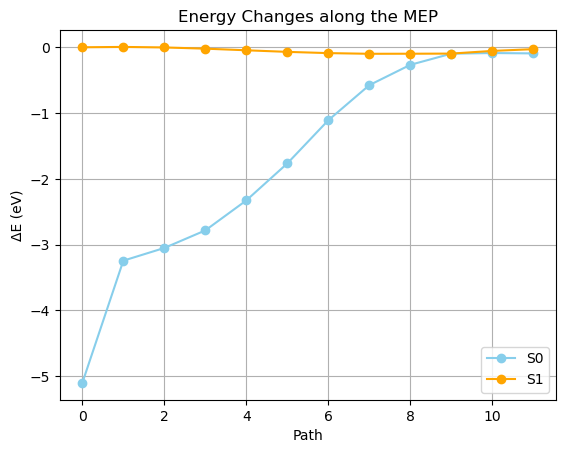
- S1状態だけをピックアップしてみると…