


この講座にぴったりの参考書 📘



量子化学計算の基礎を学ぶ 📘



量子化学計算をもっと深く学ぶ 📘



分子の安定な構造を予測できます
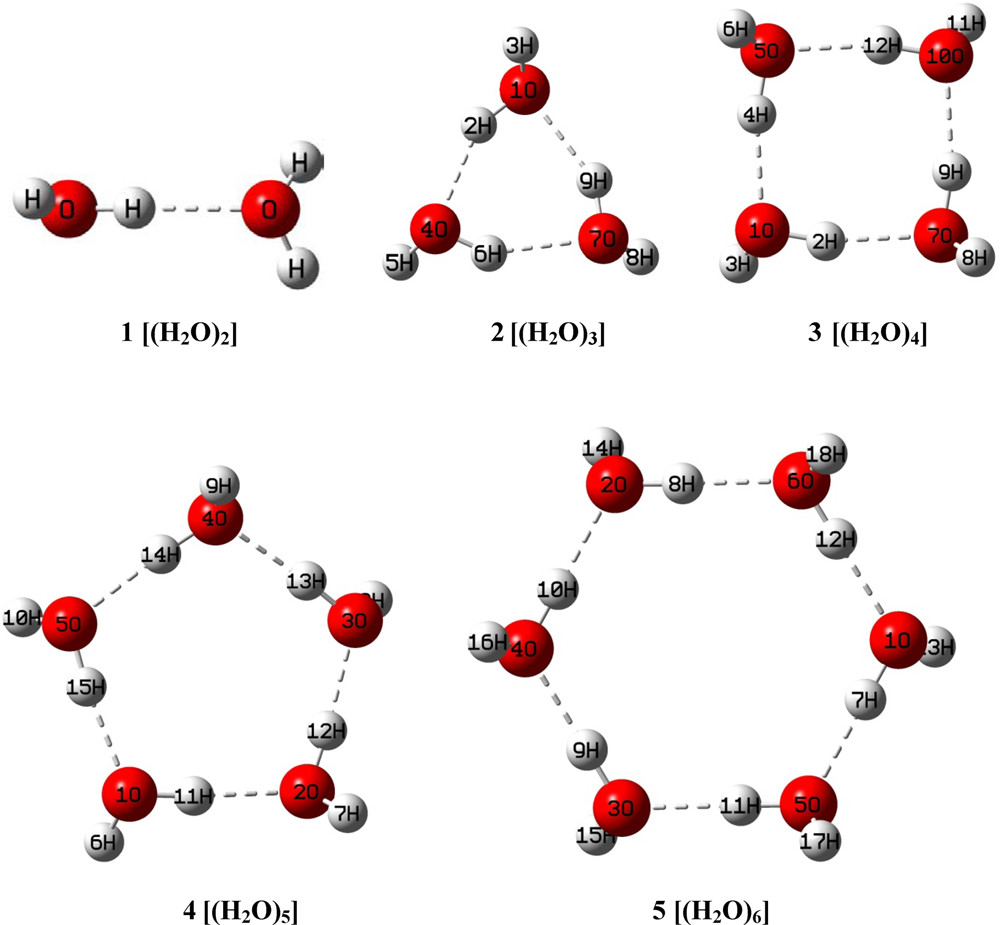
分子の振動状態を予測できます
![Solov'yov, et al. (2012)]](https://gyazo.com/48f5e3d868003070ac0fbf8dd02f1e06.png)
分子の励起状態を予測できます
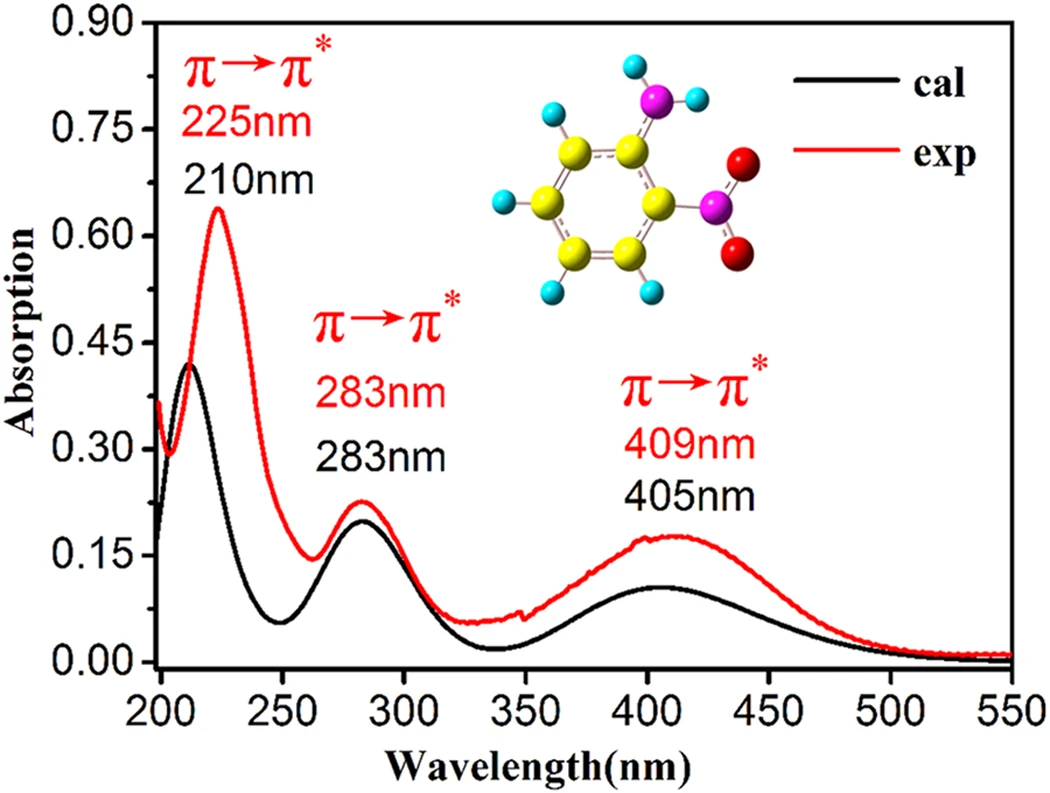
分子の電子状態に基づいて様々な解析ができます

分子の化学反応を予測できます
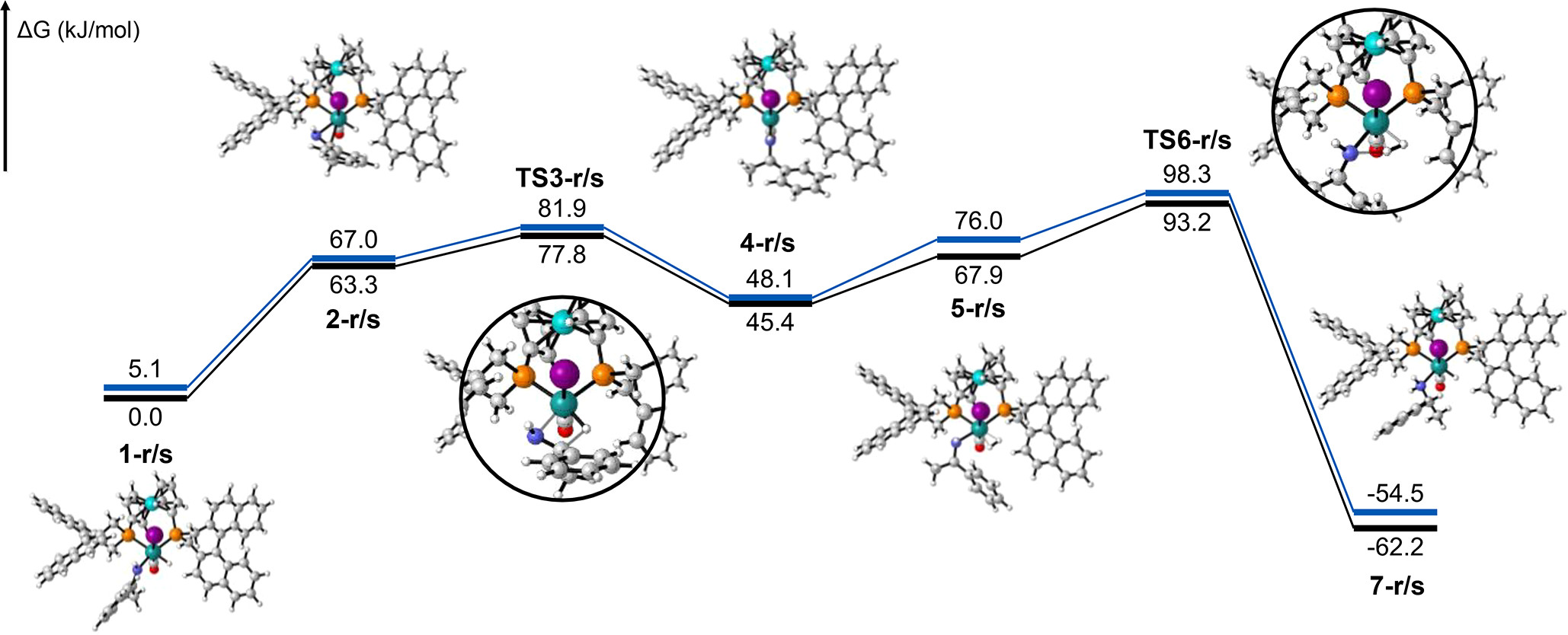
分子シミュレーション手法の比較
GaussView:ユーザーインターフェース
- GaussView というアプリと併せて使うと、高機能な量子化学計算をお手軽に実行できます
量子化学計算を体験できる Web アプリの MolCalc
- 量子化学エンジンは GAMESS
- 計算レベルは
HF/STO-3G- 計算手法:Hartree-Fock 法
- 基底関数:STO-3G
- 開発者は Copenhagen 大学の Jan Jensen ら
- 分子を SMILES 記法で指定できる
- SMILES記法とは?
- 分子の化学構造を 文字列化 して表現する表記方法
- Simplified Molecular Input Line Entry System の頭文字
- 基本的なルール
- 原子は 元素記号 で表し、水素原子 は 省略 する
- 二重結合 は「
=」、三重結合 は「#」で表す - 単結合 は(通常は)省略 する
- 環構造 は、つながっている原子の後ろに数字でラベル付け する(
C1など)- 例:プロパン:
CCC、シクロプロパン:C1CC1
- 例:プロパン:
- 芳香環 を構成する原子は小文字にする(c など)
- 例:ベンゼン:
c1ccccc1
- 例:ベンゼン:
- SMILES 記法についての詳細は こちら
| 化合物名 | 化学式 | SMILES |
|---|---|---|
| メタン | CH4 | C |
| アンモニア | NH3 | N |
| 水 | H2O | O |
| 二酸化炭素 | CO2 | O=C=O |
| 窒素 | N2 | N#N |
| 酸素 | O2 | O=O |
| エタン | C2H6 | CC |
| エチレン | C2H4 | C=C |
| アセチレン | C2H2 | C#C |
| ブタン | C4H10 | CCCC |
| 1,3-ブタジエン | C4H6 | C=CC=C |
| 1,2-ブタジエン | C4H6 C=C=CC | |
| シクロブタン | C4H8 | C1CCC1 |
| シクロブタジエン | C4H4 | C1=CC=C1 |
| シクロヘキサン | C6H12 | C1CCCCC1 |
| シクロヘキセン | C6H10 | C1CCC=CC1 |
| 1,4-シクロヘキサジエン | C6H8 | C1C=CCC=C1 |
| ベンゼン | C6H6 | c1ccccc1 |
Gaussian や GAMESS を便利に使うための WebMO
- WebMO
- Web アプリ
- ブラウザーから量子化学計算を実行できる
- 様々な量子化学計算パッケージのインターフェースとして使える
- Gaussian / GAMESS / Molpro / Mopac / NWChem / ORCA / PSI4 / Quantum Espresso / Q-Chem / VASP
- 公式サイトの デモ版
- Username:
guest - Password:
guest - 30秒までの計算が可能
- タイミングが悪いと混んでいる
- Username:
- 分子を SMILES で指定するときには、メニューから…
Lookup→Import by→SMILES- SMILES 記法
代表的な計算方法の精度とコスト

シュレディンガー方程式
$$ \left[ -\frac{1}{2} \sum_i^n \nabla_i^2 + \sum_i^n v(r_i) + \sum_{i}^n \sum_{j > i}^n \frac{1}{\left| r_{i} - r_{j} \right|} \right] \Psi = E \Psi $$
- 2項目の $v(r_i)$ は、$i$ 番目の電子に外部(原子核)からはたらくポテンシャル $$ v(r_i) = -\sum_a^{N_\mathbf{nuclei}} \frac{Z_a}{|R_a - r_i|} $$
- 3項目の $\frac{1}{|r_{i}-r_{j}|}$ は、$i$ 番目と $j$ 番目の「電子間にはたらく相互作用(電子相関)」を表す
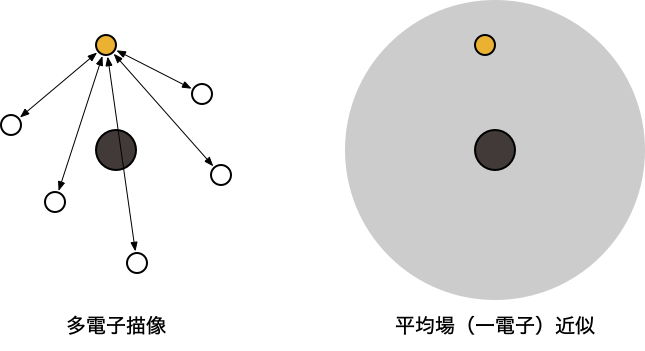
- 「ある電子は、他の電子の位置に依存しながら運動している」という描像(多電子描像)
- 厳密に取り扱うことが難しいため、何らかの「近似」が必要
- 「ある電子は、他の電子の位置に依存しながら運動している」という描像(多電子描像)
Hartree-Fock (HF) 方程式
- 量子化学計算の出発点となる最も基本的な近似
- 「ある電子は、他の電子の作る平均化された場の中で(他の電子と)独立に運動している」という描像(一電子近似)を考える
$$ \left[ -\frac{1}{2} \nabla_i^2 + v(r_i) + V_i^{\mathrm HF} \right] \psi_i = \epsilon_i \psi_i $$
- 3項目の $V_i^{\mathrm HF}$ は、$i$ 番目の電子に対して他の電子($j \neq i$)が作る「平均場」に由来する反発エネルギー
ステップ 1️⃣
- 対象とする分子系のハミルトニアン(核座標の配置 $R_\alpha$ 、核電荷 $Z_\alpha$、電子数 $n$ などの情報)、初期の参照軌道 $\Psi^{0}$ を設定する

- 対象とする分子系のハミルトニアン: $$ \hat{H} = -\frac{1}{2} \nabla_1^2 -\frac{1}{2} \nabla_2^2 - \frac{2}{r_1} - \frac{2}{r_2} + \frac{1}{r_{12}} $$
- 初期の参照軌道:
$$
\Psi^{0}(\xi_1,\xi_2)=\frac{1}{\sqrt{2}}\left[ \alpha(s_1)\beta(s_2) - \alpha(s_2)\beta(s_1)\right]\psi^{0}(r_1)\;\psi^{0}(r_2)
$$
- もちろん、ここにある $\psi^0(r_1)$ や $\psi^0(r_2)$ は、「これから求めたいもの」なので、その中身が分からない
- 拡張 Huckel 近似や半経験法など、精度の粗い方法をつかって、とりあえず求めておく
ステップ 2️⃣
- 「電子2がつくる平均場」中にある電子1について、HF 方程式を解く
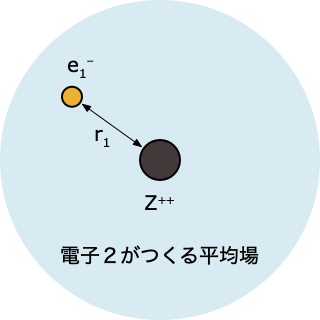
$$ \left[-\frac{1}{2}\nabla_1^2 - \frac{2}{r_1} + \int d r_2 \; |\psi^0(r_2)|^2 \, \frac{1}{r_{12}}\right]\psi(r_1)=\epsilon_1 \psi(r_1) $$
- これを解くと電子1の波動関数 $\psi(r_1)$ とエネルギー $\epsilon_1$ が求まるけれど、ステップ①で「とりあえず」ということで決めた $\psi^0(r_2)$ をつかった計算なので、精度的にはまだまだ不十分
ステップ 3️⃣
- 「電子1がつくる平均場」中にある電子2について、HF 方程式を解く
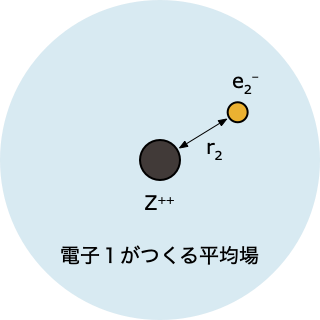
$$ \left[-\frac{1}{2}\nabla_2^2 - \frac{2}{r_2} + \int d r_1 \; |\psi^0(r_1)|^2 \, \frac{1}{r_{12}}\right]\psi(r_2)=\epsilon_2 \psi(r_2) $$
- これを解くと電子2の波動関数 $\psi(r_2)$ とエネルギー $\epsilon_2$ が求まるけれど、、ステップ①で「とりあえず」ということで決めた $\psi^0(r_1)$ をつかった計算なので、得られる答えも、精度的にはまだまだ不十分
ステップ 4️⃣
- 軌道エネルギー $\epsilon_i$ と1電子軌道 $\psi(r_i)$ から、全電子エネルギー $E$ を求める
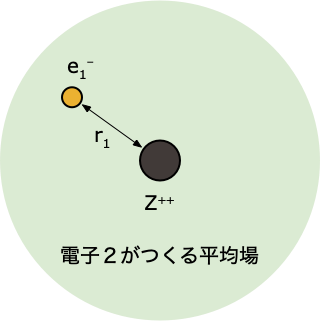
- ステップ②の続きでは、アップデートした $\psi^0(r_2)$ をつかった計算なので、得られる答えは、1回目よりも改善される(エネルギーが下がる)

- 得られた全エネルギー $E_\mathrm{new}$ と前回のもの $E_\mathrm{old}$ とのエネルギー差 $\Delta E$ を比べたときに…
- $\Delta E$ が十分に小さい
- ➡ 計算終了
- $\Delta E$ が大きい
- ➡ アップデートした軌道 $\Psi_\mathrm{new}$ を参照 $\Psi^0$ に使って、ステップ②〜④を再度実行
- $\Delta E$ が十分に小さい
電子相関とは?
- Hartree-Fock 法は、電子同士の相互作用(電子相関)を平均化して扱うという近似(平均場近似)をしている
- 電子相関の大きさは「電子相関エネルギー」($\Delta E_\mathrm{corr}$)と呼ばれ、一般に、シュレディンガー方程式の厳密解($E_\mathrm{exact}$)と HF 計算で得られる値($E_\mathrm{HF}$)との差分として定義される
- $\Delta E_\mathrm{corr} = E_\mathrm{exact} - E_\mathrm{HF}$
密度汎関数法(DFT:Density Functional Theory)とは?
- 近年、研究開発の最前線で大活躍している計算手法
- 電子密度を試行関数とする多電子状態問題の近似解法

- 電子相関の寄与について、 Hartree-Fock 法とほぼ同程度の計算コストで考慮することができる
- Hartree-Fock 法 などは波動関数を試行関数とする方法であるが、DFT は電子密度を試行関数とする方法であり、その基盤となる考え方が大きく異なっている
- HF 法の基礎は HF 方程式だが、DFTを実践する上で鍵になるのは コーン・シャム(Kohn-Sham; KS)方程式
Kohn-Sham 系のイメージ

Gaussian で利用できる様々な汎関数

交換・相関汎関数の大まかな分類
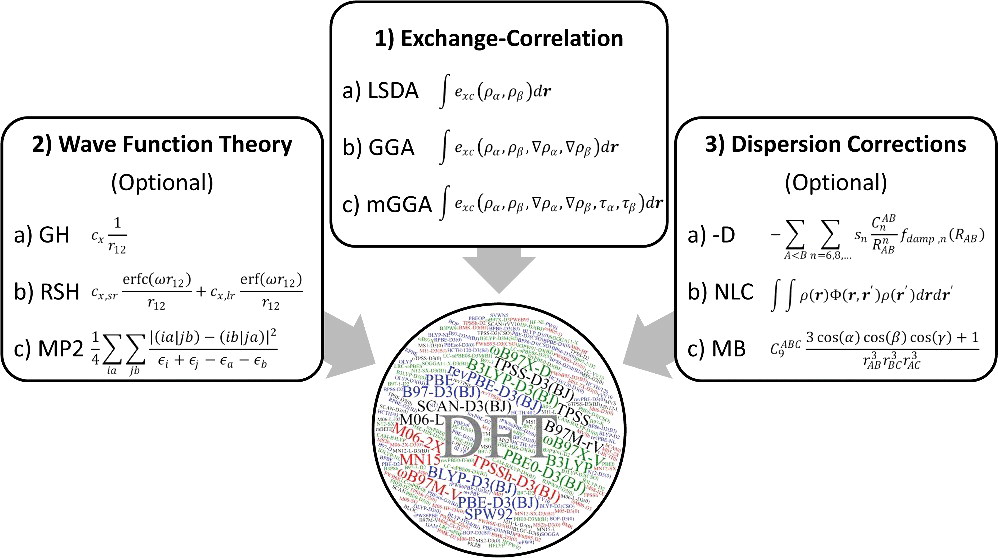
交換・相関汎関数の種類と計算精度

交換・相関汎関数の階層的な分類
- Perdew によって提案された汎関数の階層的な分類です
- Jacob のはしご (Wikipedia) 、と呼ばれています

交換・相関汎関数の例
- 代表的な200個の交換・相関汎関数をまとめると…
Local:非 hybrid 型Disp:分散力補正あり下線:長距離補正あり
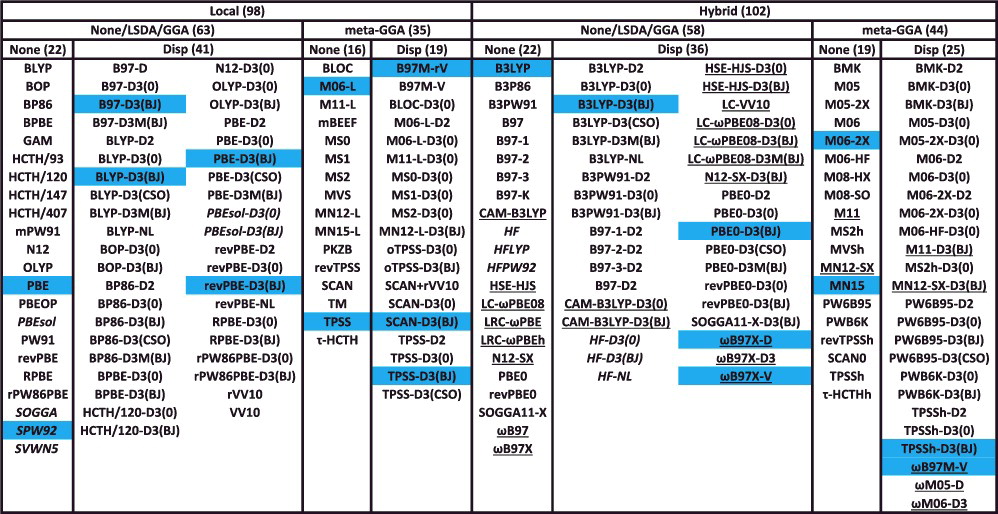
hybrid 交換・相関汎関数 📊
- hybrid 型は、LSDA / GGA / mGGA などの交換汎関数に対して、一定の割合で Hartree-Fock 交換積分 $E_\mathrm{x}^\mathrm{HF}$ を混合した汎関数です
- 非 hybrid 型(local 型)と hybrid 型の汎関数ペア(例:BLYP vs B3LYP)について、計算される物性値の平均二乗偏差(RMSD)を比較してみると…
NCD:分子間相互作用エネルギーID:異性化エネルギーBH:反応障壁の高さ
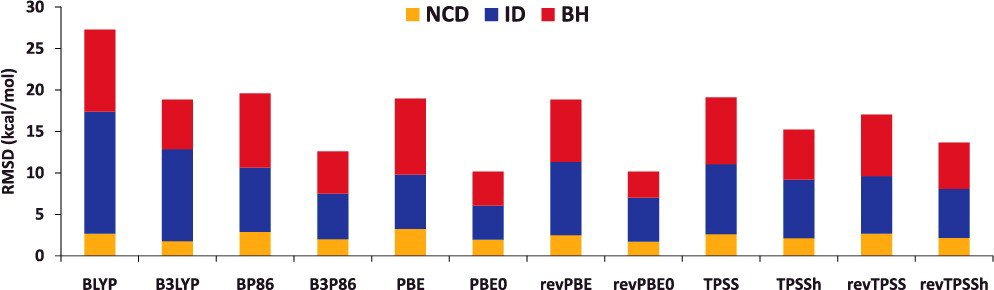
- $E_\mathrm{x}^\mathrm{HF}$ を適度に hybrid することで計算精度が向上
分散力補正 📊
- 標準的な DFT の弱点のひとつは、分散力(van der Waals 相互作用) の記述ができない
- このために、分子集合体における分子間相互作用を過小評価する傾向にある
- Grimme は、分子力場などで用いられるように経験的なパラメータを用いて、各原子対に対する引力的なエネルギー項を付加する分散力補正を提案
- Grimme の分散力補正のあり・なし(例:BLYP と BLYP-D2, BLYP-D3)について、計算される物性値の平均二乗偏差(RMSD)を比較してみると…
NCED:分子間相互作用エネルギーIE:異性化エネルギー
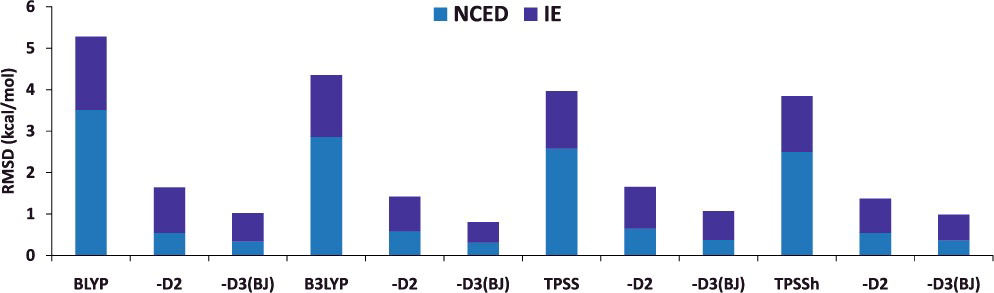
- Grimme の分散力補正を加えることで、コストを追加することなく、計算精度を向上させることができる
交換・相関汎関数のベンチマーク 📊
- 200 個の汎関数について、ベンチマーク計算をしてみると…
- 汎関数名の右にある数字は、計算精度のランキング
- 左にあるラベルは、評価した物性値:
NCED/NCED/NCD:分子間相互作用エネルギーIE/ID:異性化エネルギーTCE/TCD:熱化学量BH:反応障壁の高さ

- GGA から meta GGA に変えても、多くの場合、計算精度はそれほど良くはならない
- 非 hybrid 型(local 型)から hybrid 型に変えると、ほとんどの場合、計算精度が向上
Gaussian で利用できる様々な基底関数

展開係数とは?
- 量子化学計算では、あらかじめ適切にパラメータを設定した基底関数 ${\phi_n}$ を使って、展開係数 ${c_n}$ の値について、最適なものを探そうとする
- つまり、計算の途中で…
- 基底関数 ${\phi_n}$ は変えずに、
- 展開係数 ${c_n}$ の値だけを変える。

水素分子の場合
- 水素分子を表す波動関数 $\Psi$ は、たとえば、2個の水素原子 $\mathrm{H}_a$ と $\mathrm{H}_b$ の中心に置いた1s 原子軌道 $\phi_a$ と $\phi_b$ の線形結合として、次のように表すことができる $$ \Psi = c_a , \phi_a + c_b , \phi_b $$
- 水素原子については、1s 軌道の厳密解が次のように分かっている $$ \psi_\mathrm{H1s} = \sqrt{\frac{1}{\pi}} \exp(-r) $$
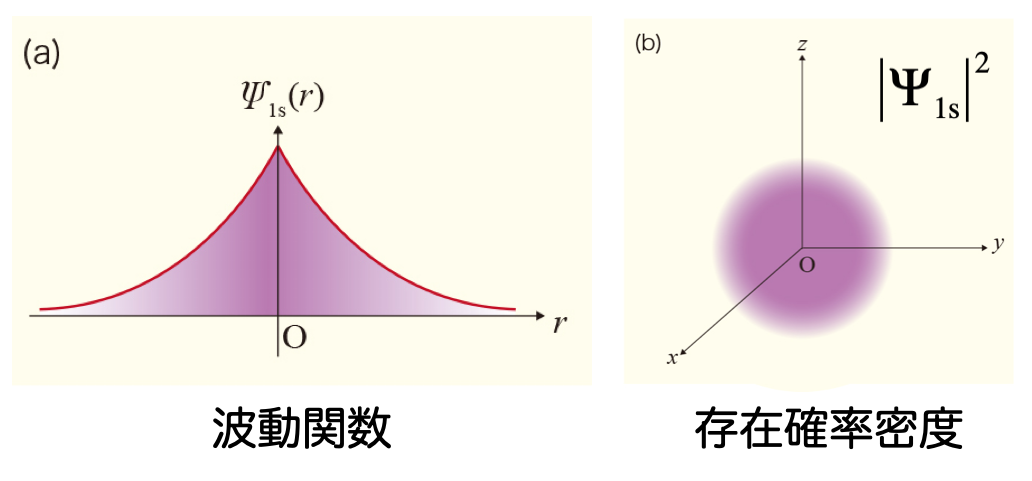
- したがって、水素原子の 1s 軌道を参考にして、$\exp(-\zeta r)$ 型の関数を基底関数として使っても良さそうだ
$$ \phi_a = \exp(-\zeta r) $$
- このような関数をスレーター型関数 と呼ぶ
- 係数 $\zeta$ は、関数の「広がりの程度」を表す変数
- 水素原子の厳密な解析解(= 電子の分布を適切に表すことができる)なので、計算精度は高い

- ただし、数値計算の計算効率は悪い
- 図にあるように、原子の中心($r = 0$)で値が不連続になるから、数値的に積分することが難しい
- 計算効率が悪いので、ふつうは、量子化学計算では使わない
ガウス型関数
- 量子化学計算では、スレーター型関数 $\exp(-\zeta r)$ の代わりに、$\exp(-\alpha r^2)$ 型の関数を基底関数として使う
$$ \phi_a = \exp(-\alpha r^2) $$
- このような関数を ガウス型関数 と呼ぶ
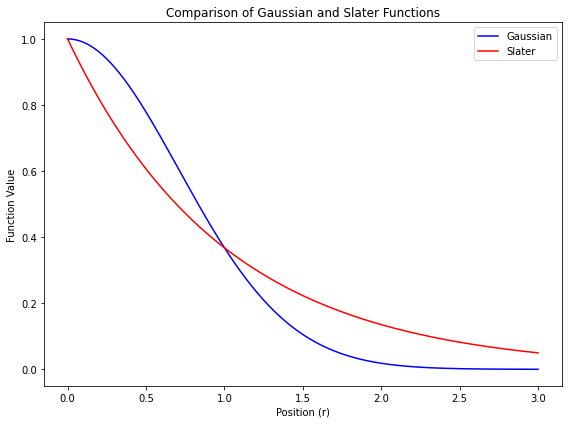
- 係数 $\alpha$ は、関数の「広がりの程度」を表す変数
- 量子化学計算の前に、あらかじめ設定されている定数(計算の途中で変化させない)
- ガウス型関数は、原子軌道に対する近似解なので、計算精度は高くはない
- ただし、数値計算の計算効率が良い
- 上の図にあるように、原子の中心($r = 0$)でも値が連続になるから、数値積分が簡単
- 計算精度の悪さは、拡がりの程度を変えたガウス型の基底関数をいくつか線形結合させることでカバーする
- いくつかのガウス型関数を線形結合させたものを# 短縮型ガウス基底とよぶ
- たとえば…
$$ \phi_\mathsf{blue} = c_\mathsf{cyan} \, \chi_\mathsf{cyan} + c_\mathsf{pink} \, \chi_\mathsf{pink} + c_\mathsf{green} \, \chi_\mathsf{green} $$ $$ = \left(1.33 \; e^{-0.59 \; r^2_\mathsf{cyan}}\right) + \left(-0.57 \; e^{-1.28 \; r^2_\mathsf{pink}}\right) + \left(0.04 \; e^{+0.06 \; r^2_\mathsf{green}}\right) $$
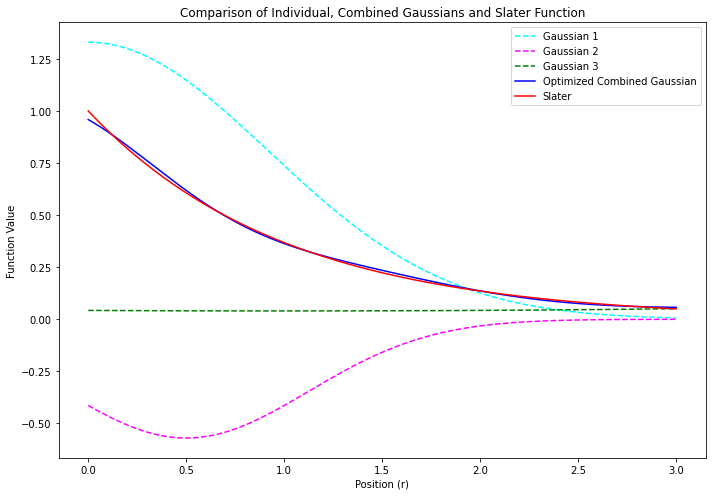
- 青い線で示す 短縮型ガウス関数 は、赤い線で示すスレーター型関数のかたちを、まあまあ良く再現している
分割価電子(split valence)基底関数
- 内殻軌道には1つ、価電子軌道には複数の短縮ガウス型関数を用いた基底関数系
- Pople 系の場合
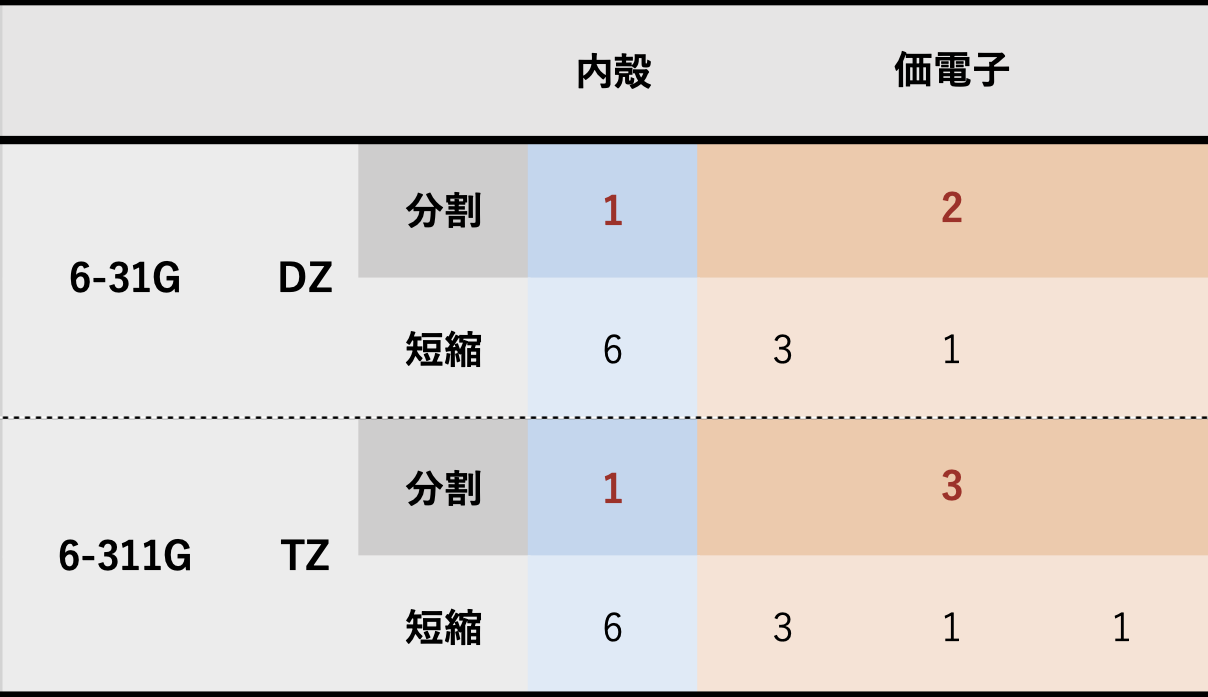
具体例

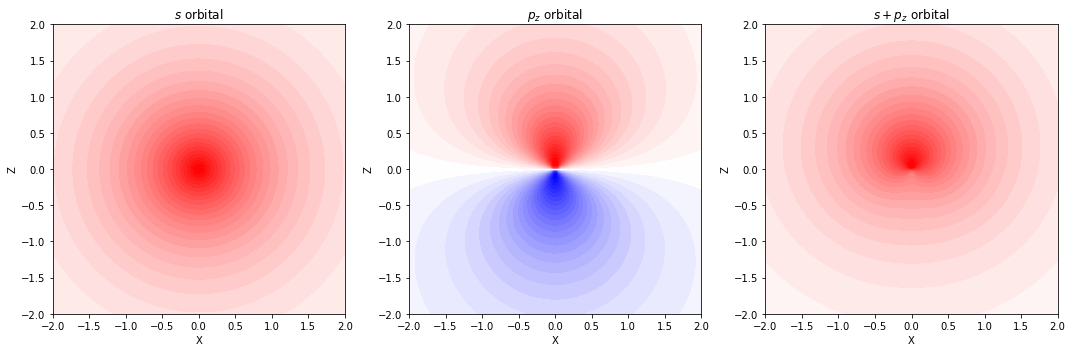
分極関数(d 型関数)
- p 軌道をもつ水素以外の原子に対しては、d 型の関数を追加する
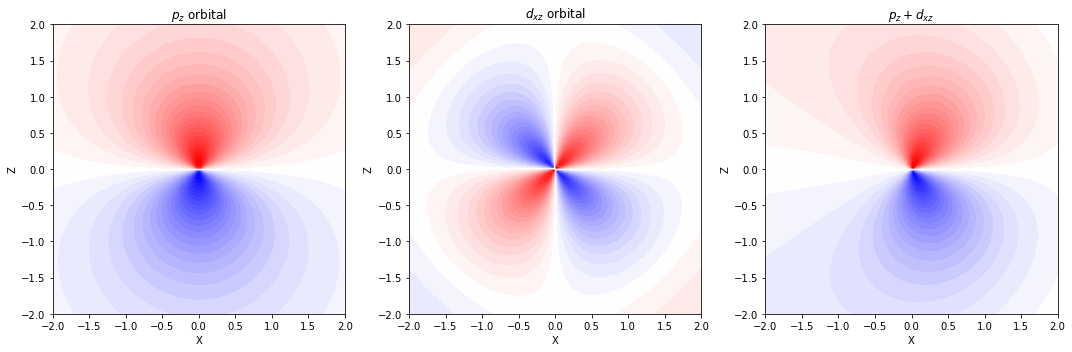
分極関数(p 型関数)
- 球対称な s 軌道のみを持つ水素原子に対しては、p 型の関数を追加する
Gaussian で分極関数を追加するには

Gaussian で分散関数を追加するには

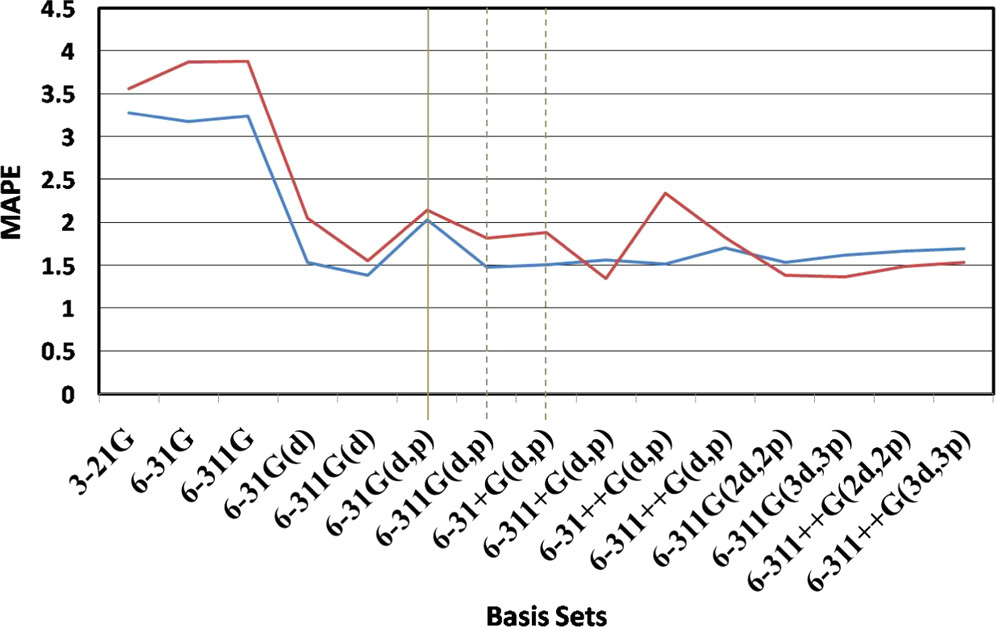
基底関数のベンチマーク(VSCF-PT2 計算の場合) 📊
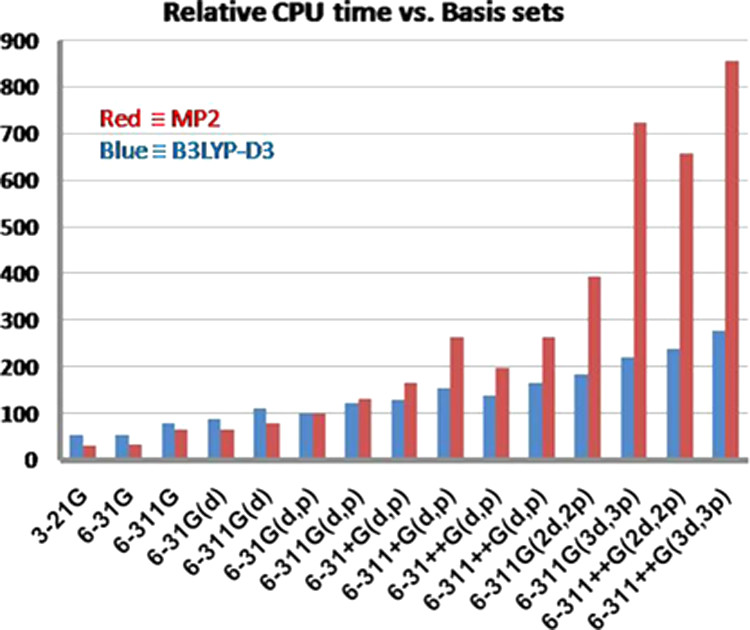
基底関数と計算時間(VSCF-PT2 計算の場合) 📊
DFT 汎関数と基底関数の組み合わせ

分子の安定構造の探索
- 各原子核に働く力(エネルギー勾配 $\frac{\partial E}{\partial Q}$)の方向にカタチをちょっとずつ変化($x_\mathrm{new} = x_\mathrm{old} - \alpha \frac{\partial E}{\partial Q}$)させることで、分子はより安定となる( = エネルギーが小さくなる)
- $E(x_\mathrm{new}) < E(x_\mathrm{old})$

- 「カタチをちょっとずつ変化させる」を繰り返すことで、分子の立体構造を半自動的に最適化できる
- 実際には、多くの量子化学計算プログラムは、エネルギー勾配の方向に動かすという単純な方法(勾配降下法)ではなく、もっと効率的なアルゴリズムを使っている
- Gaussian では、デフォルトでは、GEDIISというアルゴリズムを用いる
- 実際には、多くの量子化学計算プログラムは、エネルギー勾配の方向に動かすという単純な方法(勾配降下法)ではなく、もっと効率的なアルゴリズムを使っている
計算例:水分子の構造最適化

分子振動とは?
- 水($\mathrm{H_2O}$)の変角振動 / 対称伸縮振動 / 逆対称伸縮振動
赤外吸収スペクトル 📈 とは?

計算例:水分子の振動解析

Gaussian で熱力学量を調べる

熱力学補正の具体例
- $\mathsf{C_2H_5 + H_2 \rightarrow C_2H_6 + H}$
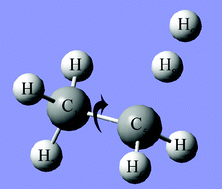
- エネルギー変化(熱力学補正をしない場合)
- $\Delta E = E_\mathsf{生成物} - E_\mathsf{反応物}$ = 4.86 kcal/mol
- 反応エンタルピー
- $\Delta_r H^\circ(298\mathsf{K}) = H_\mathsf{生成物} - H_\mathsf{反応物}$ = 8.08 kcal/mol
- Gibbs 自由エネルギー変化
- $\Delta_r G^\circ(298\mathsf{K}) = G_\mathsf{生成物} - G_\mathsf{反応物}$ = 11.18 kcal/mol
分子振動の非調和性
- 高い精度(MP2/cc-pVTZ)で計算した場合

- MP2摂動法を用いることで、実験値にかなり近くなる
- 高波数の振動モード(対称伸縮&逆対称伸縮)では、誤差が大きい
- 分子振動を 調和振動子 だと見なす近似(調和近似)による誤差(非調和性)の影響だと考えられる
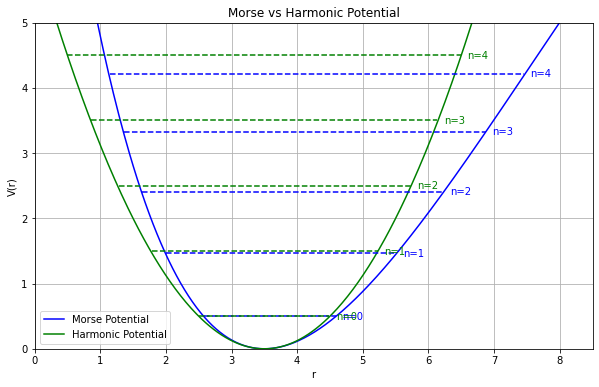
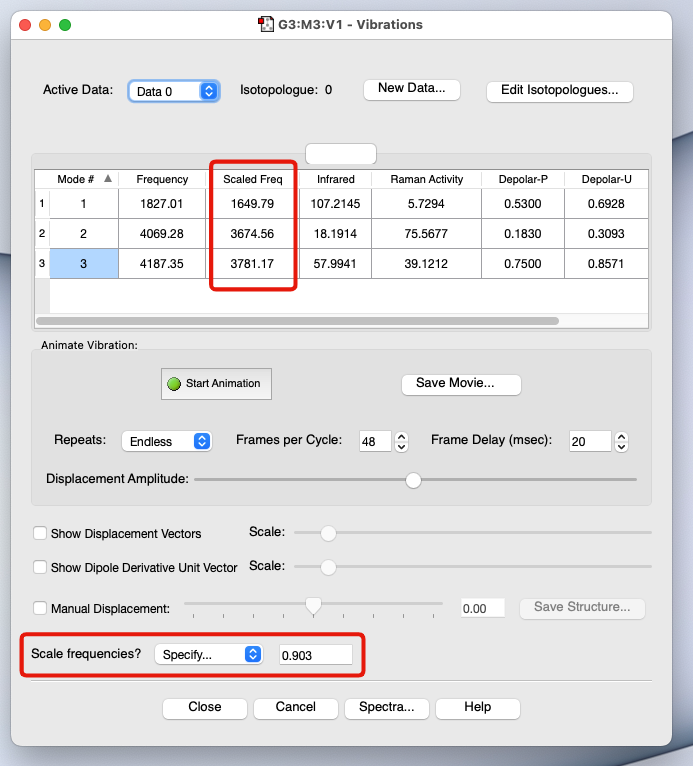
スケーリング因子
- 非調和性の問題を手軽に解決する場合には、振動数に適当な数字(スケーリング因子)を掛けて補正する
- 参考: https://cccbdb.nist.gov/vibscalejust.asp
- 事例:
HF/6-31G(d)の場合 → 0.903B3LYP/6-31G(d,p)の場合 → 0.961
非調和振動数解析
- 分子振動をより定量的精度で解析したい場合には、調和近似を越えた取り扱いが必要
- GAMESS では、非調和性を考慮した振動解析として、Vibrational SCF (VSCF) 法を利用することができる
- 詳細は こちら
- VSCF法で計算した場合

💻 実演:Gaussian を用いた Diels-Alder 反応の解析
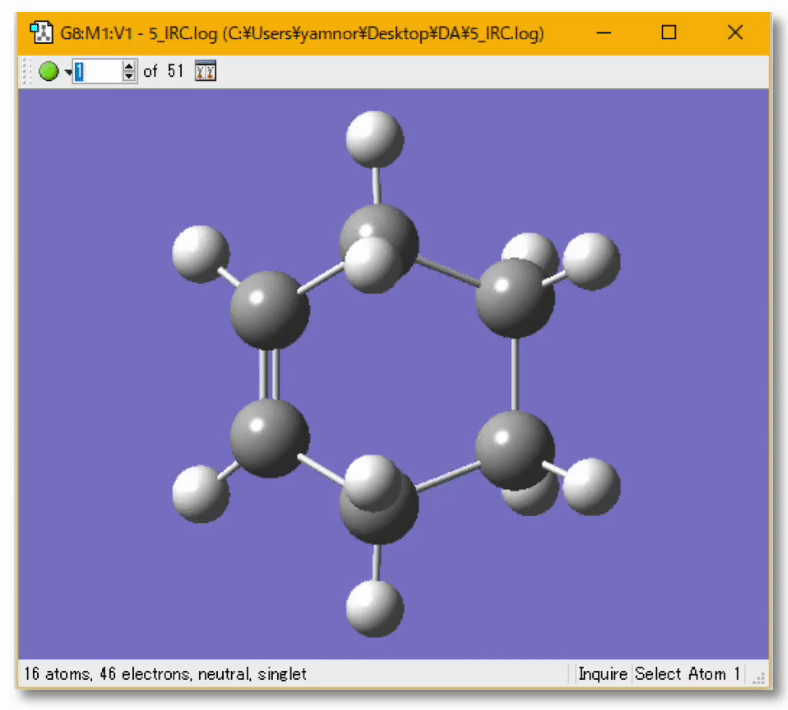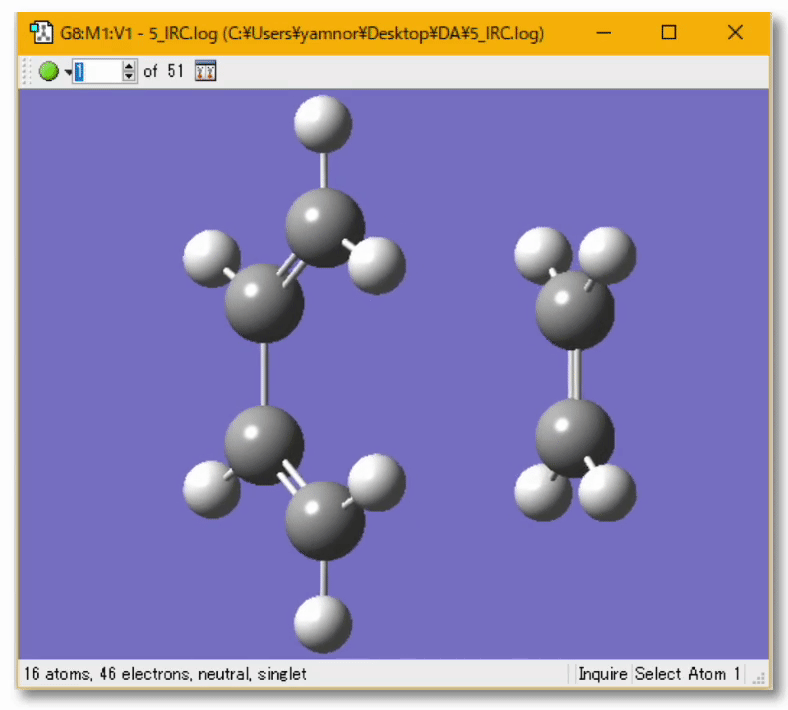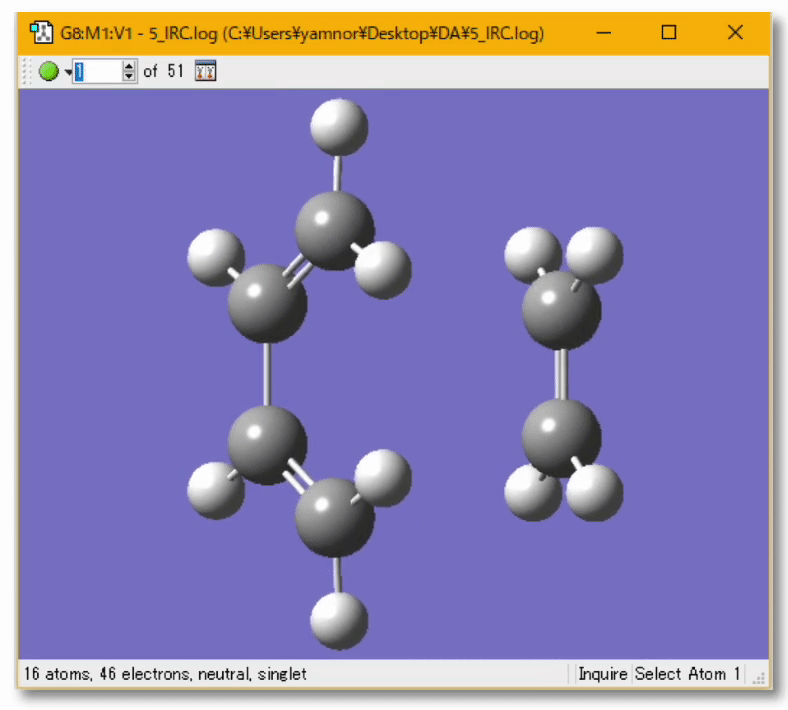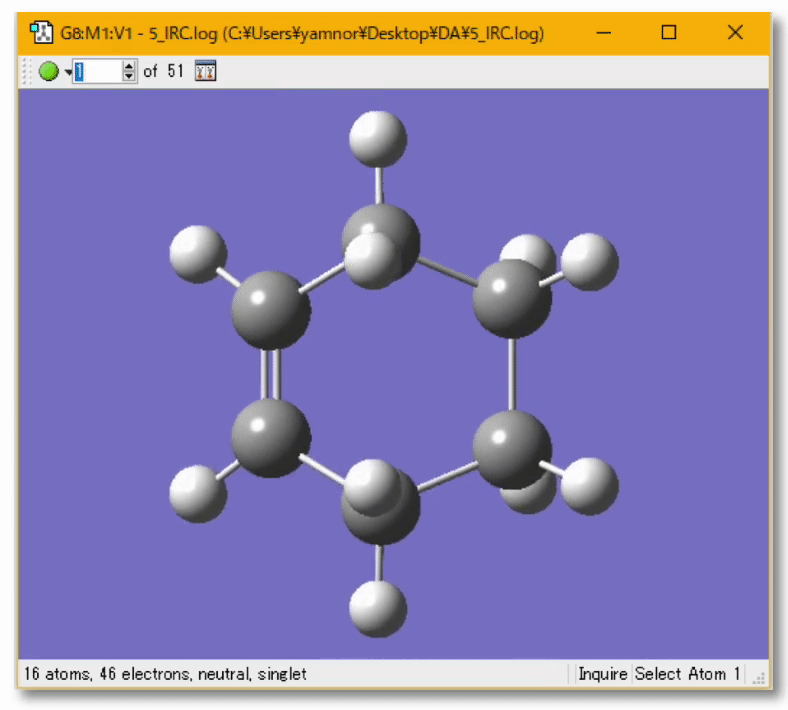
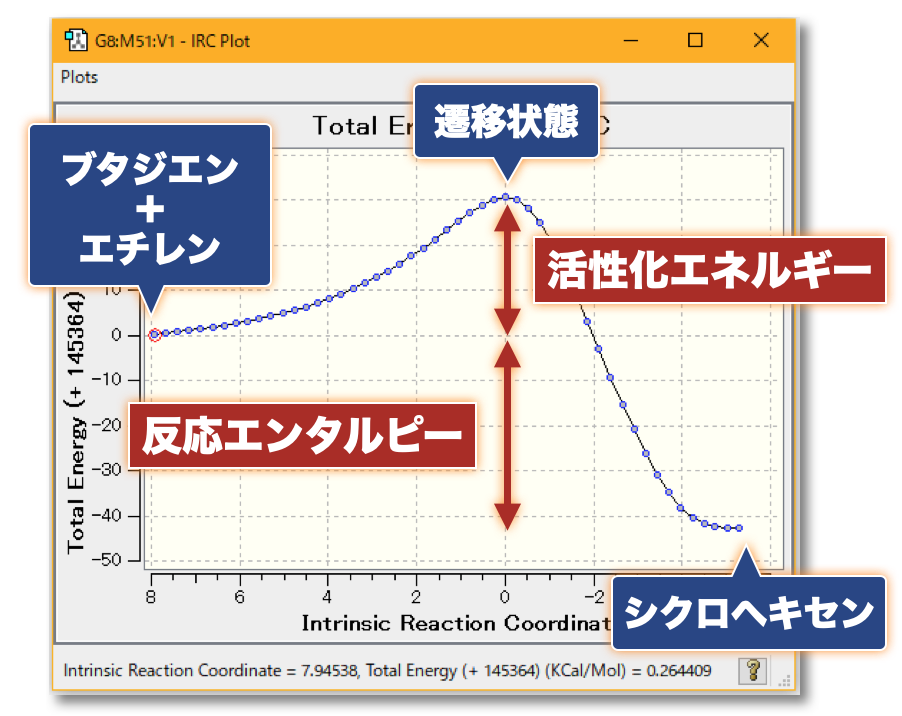
分子の遷移状態
- ある化学反応について、反応速度を議論するときには、その反応に伴う 遷移状態(Transition State; TS) を調べる必要がある
- たとえば、ある反応の速度定数 $k$ について、始状態と遷移状態とのエネルギー差、つまり、活性化エネルギー $\Delta G^{\ddagger}$ が分かっていれば、Eyring-Polanyi 式から…
$$ k = \chi \frac{k_\mathrm{B}T}{h}\exp\left(-\frac{\Delta G^{\ddagger}}{RT}\right) $$- $\chi$:遷移状態において、反応座標に沿った分子振動が一往復したときに反応する確率(透過係数)
- $h$: プランク定数
遷移状態の探索
- 多くの量子化学計算プログラムには、遷移状態を探索する機能が備わっている
- 単純に「構造最適化の逆」($x_\mathrm{new} = x_\mathrm{old} + \alpha \frac{\partial E}{\partial Q}$)をやっても、うまくいかない
- エネルギーの二次微分($\frac{\partial^2 E}{\partial Q^2}$)を要素にもつ Hessian 行列 の固有値(力の定数 or 曲率:$\chi$)を求めて…
- Hessian 行列の負の固有値を持つ固有ベクトルの方向に少しずつ動かすという方法がある
- → Eigenvector Following 法
- 調和近似($E = \kappa Q^2$)からの歪みが大きな方向に少しずつ動かすという方法がある
- → 超球面探索法(参考: GRRMプログラム )
- Hessian 行列を計算するためのコストがまあまあ大きい
- ➡ 構造最適化よりも計算コスト(時間)がかかる
- Hessian 行列の負の固有値を持つ固有ベクトルの方向に少しずつ動かすという方法がある
💻 実演:Gaussian を用いたブタジエンの cis/trans 異性化反応の解析
- 遷移状態に近い(と予想される)構造から出発する
- 今回の場合、C-C 軸周りの回転角度を 90度くらいにしてみる
- 見つけようとしている遷移状態は捻れ型だと考えられるから
- 今回の場合、C-C 軸周りの回転角度を 90度くらいにしてみる
- 遷移状態探索の設定方法
Job Type
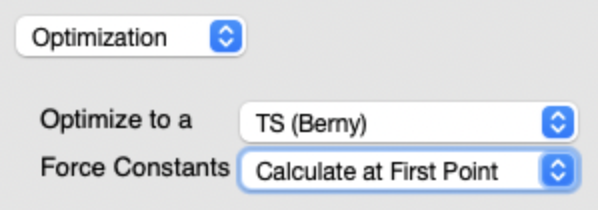
Optimizationを選択Optimize to aTS (Berry)Force ConstantsCalculate at First Point
化学反応のエネルギーダイアグラムを描く

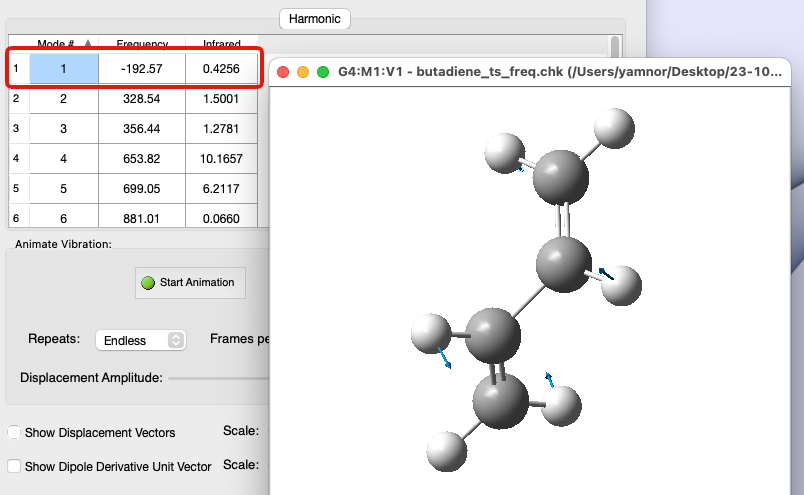
遷移状態で振動解析すると…
- 値が負(虚数)となる振動数がある
安定構造では…
- エネルギーの座標に関する一次微分(エネルギー勾配)が常にゼロである $$ \frac{\partial E}{\partial Q} = 0 $$
- 局所 安定状態(下に凸の放物線) であり、エネルギーの座標に関する二次微分の値が常に正 である $$ \frac{\partial^2 E}{\partial Q^2} > 0 $$
光の吸収とは?
- 光のエネルギーが分子を基底状態から励起状態に変えるエネルギーとして消費されるプロセス

- $E_{励起} - E_{基底} = \Delta E = h c / \lambda_{abs}$ に相当する光のエネルギーを分子が吸収し、基底 → 励起状態への電子遷移が起こる
- $h$: Planck 定数
- $c$:光の速度
分子の吸光・発光と基底・励起状態

- 吸光
- 基底($\mathrm{S_0}$)状態の最適構造 → 励起($\mathrm{S_n}$)状態
- つまり、量子化学計算に取り組むときに、分子の立体構造は、基底状態で最適化する必要がある
- 基底($\mathrm{S_0}$)状態の最適構造 → 励起($\mathrm{S_n}$)状態
- 発光
- 励起($\mathrm{S_n}$)状態の最適構造 → 基底($\mathrm{S_0}$)状態
- つまり、量子化学計算に取り組むときに、分子の立体構造は、励起状態で最適化する必要がある
- 励起($\mathrm{S_n}$)状態の最適構造 → 基底($\mathrm{S_0}$)状態
Gaussian で TD-DFT を用いて励起状態を計算する

TD-DFT のベンチマーク(汎関数依存性) 📊

- 領域分割 hybrid 型(長距離補正)を用いることで、計算精度が改善しています。
- 蛍光(fluorescence)の結果に関しては、ベンチマークに用いたデータが少ないので、信頼性に欠けるかもとのこと。
溶媒和とは
- 溶質分子が、静電気力や水素結合などによって結びつき取り囲むことで溶質が溶媒中に拡散する現象
溶媒効果とは?
- 化学において、反応性もしくは分子の会合に対して溶媒が及ぼす影響を指す
- 溶媒は溶解度、安定性、反応速度に影響を及ぼすため、適切な溶媒を選択することにより、化学反応を熱力学的・速度論的に制御できる
- 溶媒効果の例
- アセチルアセトンの互変異性に対する平衡定数: $$ K_\mathrm{keto-enol} = \mathrm{\frac{[enol]}{[keto]}} $$

- ある可逆的な反応過程 $A$ について、対応する平衡定数 $K$ とギブス自由エネルギー変化 $\Delta G$ の関係
$$ \Delta G = -RT \ln K $$
- $R$:気体定数($= k_\mathrm{B} N_\mathrm{A} = 8.314,462 \; \mathrm{J \; K^{-1} \; mol^{-1}}$)
- $T$:温度(300 K)
| 溶媒の種類 | keto-enol 平衡定数 | ギブス自由エネルギー変化 |
|---|---|---|
| 気相 | 11.7 | -6.12 |
| 水 | 0.23 | 3.67 |
| ジクロロメタン | 4.2 | -3.58 |
| エタノール | 5.8 | -4.39 |
| テトラヒドロフラン | 7.2 | -4.93 |
| ベンゼン | 14.7 | -6.70 |
| シクロヘキサン | 42 | -9.32 |
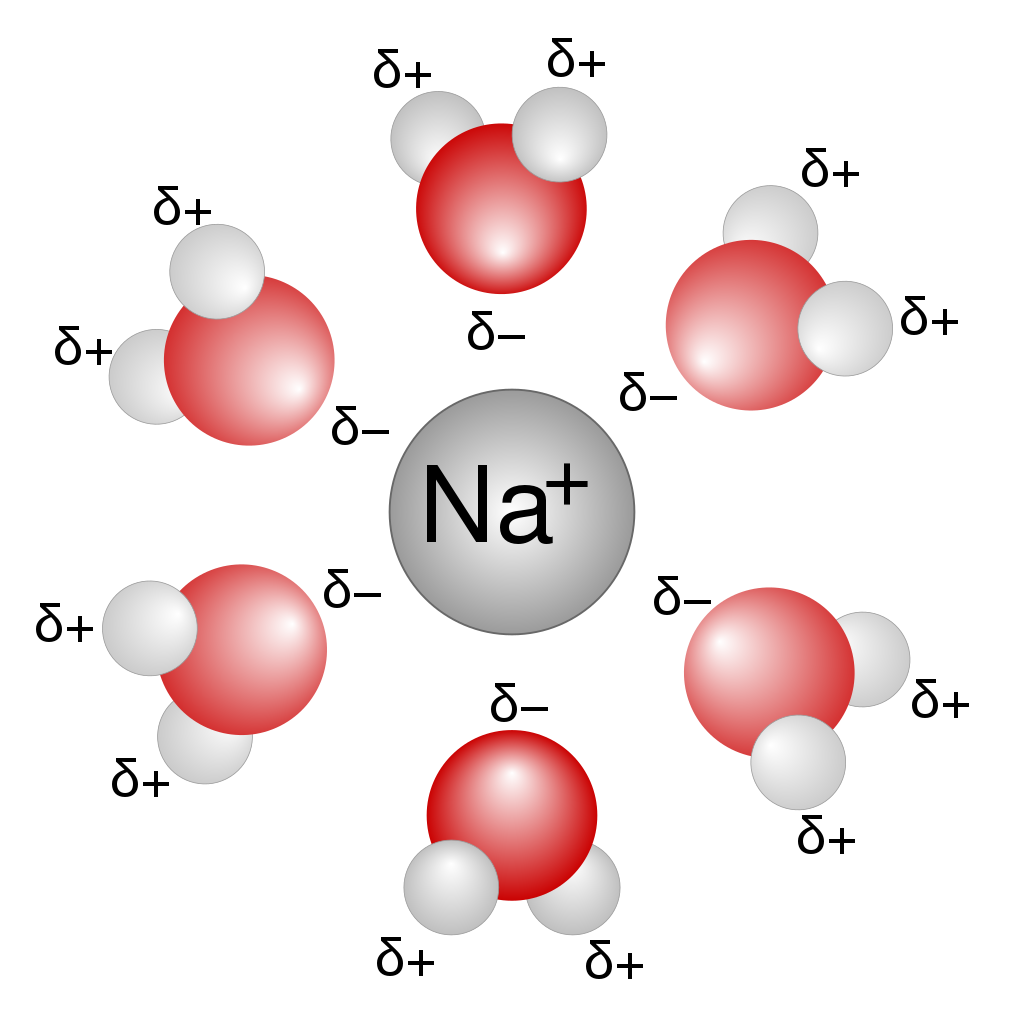
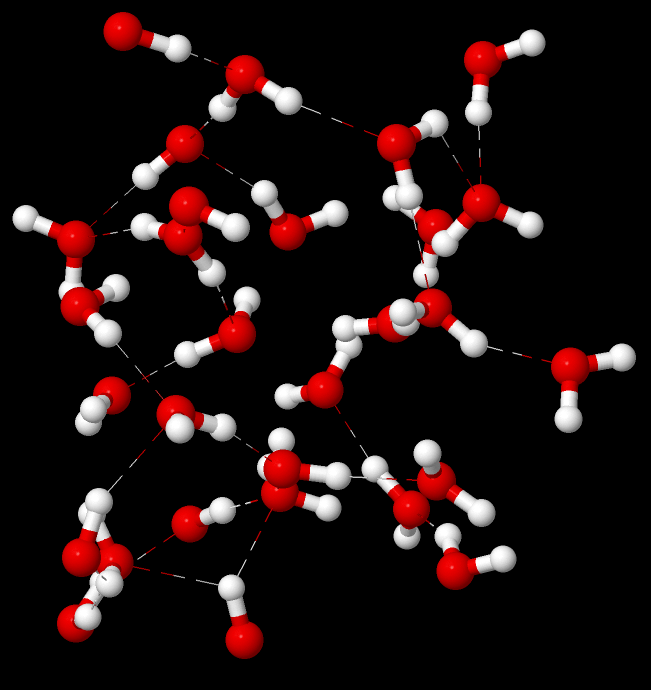
溶媒分子を露わに考慮する(explicit solvation model)
溶媒分子を分極する連続誘電体として扱う(implicit solvation model)
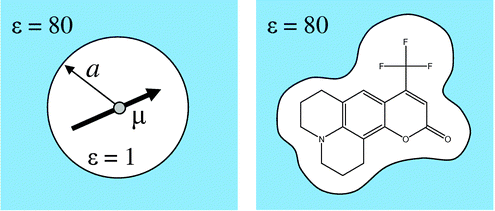
- 左:Debye-Onsager モデル
- 右:連続誘電体モデル
Gaussian で溶媒効果を考慮する



